The soluble guanylate cyclase activator cinaciguat prevents cardiac dysfunction in a rat model of type-1 diabetes mellitus
█ Original article
DOI: 10.26430/CHUNGARICA.2017.47.1.34
Authors:
Mátyás Csaba1*, Barta Bálint András1*, Németh Balázs Tamás1, Oláh Attila1, Hidi László1, Birtalan Ede1, Kellermayer Dalma1, Ruppert Mihály1, Korkmaz-Icöz Sevil2, Kökény Gábor3, Horváth Eszter Mária4, Szabó Gábor2, Merkely Béla1, Radovits Tamás1
1Semmelweis Egyetem, Városmajori Szív- és Érgyógyászati Klinika, Kísérleti Kutató Laboratórium, Budapest
2Ruprecht-Karls Egyetem, Szívsebészeti Klinika, Kísérleti Kutató Laboratórium, Heidelberg, Németország
3Semmelweis Egyetem, Kórélettani Intézet, Budapest
4Semmelweis Egyetem, Élettani Intézet, Budapest
*A szerzők a munkához egyenlő mértékben járultak hozzá.
Summary
Introduction: Diabetes mellitus (DM) leads to the development of diabetic cardiomyopathy, which is associated with altered nitric oxide (NO) – soluble guanylate cyclase (sGC) – cyclic guanosine monophosphate (cGMP) signaling. Cardio-protective effects of elevated intracellular cGMP-levels have been described in different heart diseases. In the current study we aimed at investigating the effects of pharmacological activation of sGC in diabetic cardiomyopathy.
Methods: Type-1 DM was induced in rats by streptozotocin. Animals were treated either with the sGC activator cinaciguat (10 mg/kg/day) or with placebo orally for 8 weeks. Left ventricular (LV) pressure-volume (P-V) analysis was used to assess cardiac performance. Additionally, gene expression (qRT-PCR) and protein expression analysis (Western blot) were performed. Cardiac structure, markers of fibrotic remodelling and DNA damage were examined by histology, immunohistochemistry and TUNEL assay, respectively.
Results: DM was associated with deteriorated cGMP signalling in the myocardium (elevated phosphodiesterase-5 expression, lower cGMP-level and impaired PKG activity). Cardiomyocyte hypertrophy, fibrotic remodeling and DNA fragmentation were present in DM that was associated with impaired LV contractility and diastolic function. Cinaciguat treatment effectively prevented DM related molecular, histological alterations and significantly improved systolic and diastolic function.
Conclusions: Cinaciguat prevented structural, molecular alterations and improved cardiac performance of the diabetic heart. Pharmacological activation of sGC might represent a new therapy approach for diabetic cardiomyopathy.
ISSUE: CARDIOLOGIA HUNGARICA | 2017 | VOLUME 47, ISSUE 1
Összefoglalás
Bevezetés: A cukorbetegség diabéteszes cardiomyopathia kialakulásához vezet, amely károsodott nitrogén-monoxid (NO) – szolubilis guanilát-cikláz (sGC) – ciklikus guanozin-monofoszfát (cGMP) jelátvitellel társul. A megnövelt intracelluláris cGMP-szint kardioprotektív hatását több szívbetegségben is leírták. Jelen kísérletünkben az sGC farmakológiai aktivációjának hatását vizsgáltuk diabéteszes cardiomyopathiában.
Módszerek: Patkányainkban 1-es típusú diabetes mellitust (DM) streptozotocinnal indukáltunk. Ezt követően az állatok per os sGC-aktivátor cinaciguatot (10 mg/testtömeg kg/nap) vagy placebót kaptak 8 héten át. A kardiális funkció megítélésére bal kamrai (BK) nyomástérfogat (P-V) analízist végeztünk. Gén- (qRT-PCR) és proteinexpressziós (Western-blot) vizsgálatokat hajtottunk végre BK-i szívizomszövetből. A myocardium strukturális változását, fibrotikus átépülésének jeleit és a DNS-károsodás mértékét szövettani- és immunhisztokémiai vizsgálatokkal tanulmányoztuk.
Eredmények: Cukorbetegségben károsodott miokardiális cGMP-jelátvitelt láttunk (emelkedett foszfodiészteráz-5 expresszió, csökkent cGMP-szint és protein-kináz-G aktivitás). Emellett szívizomsejt-hipertrófia, fibrotikus átépülés és DNS-töredezettség volt jelen a BK-ban, ami romlott kontraktilitással és diasztolés diszfunkcióval párosult. A cinaciguat kezelés hatásosnak bizonyult a DM indukálta molekuláris és szövettani eltérések megelőzésében, továbbá szignifikánsan javította a szisztolés és a diasztolés funkciót DM-ben.
Következtetések: A cinaciguat megelőzte a diabéteszes szív strukturális és molekuláris változásait, illetve javította annak pumpafunkcióját. Mindezek alapján az sGC farmakológiai aktivációja új terápiás megközelítést képviselhet a diabéteszes cardiomyopathia kezelésében.
Bevezetés
A diabetes mellitus (DM) olyan kardiovaszkuláris komplikációkkal mutat összefüggést, mint a miokardiális infarktus, krónikus szívelégtelenség, illetve különböző vaszkuláris megbetegedések. Ugyanakkor diabéteszben a megváltoz ott metabolizmus a szívizomzatra kifejtett hatása eredményeként egy speciális betegség, a diabéteszes cardiomyopathia alakul ki (1). Számos irodalmi adat ellenére (szisztolés, diasztolés diszfunkció, miokardiális fibrózis és hipertrófia, nitro-oxidatív stressz, gyulladás, apoptózis) (2–4) a diabéteszes cardiomyopathia kialakulásának mechanizmusa még nem ismert (1, 5–7).
Élettani körülmények között az endothel nitrogén-monoxid (NO) szintáz enzime (eNOS) NO-t termel, amely átdiffundálva a simaizom- és szívizomsejtekbe, intracelluláris receptorát, a szolubilis guanilát-cikláz enzimet (sGC) aktiválja. Az sGC ennek hatására a másodlagos hírvivő ciklikus guanozin-monofoszfát (cGMP) termelésébe kezd, majd a cGMP aktiválja a cGMP-függő protein-kináz G-t (PKG), amely a jelátviteli út legfőbb effektora (8). DM-ben az NO-sGC-cGMP-PKG-jelátvitel zavarának számos mechanizmusát írták le úgy, mint a reaktív nitrogén- és oxigénszármazékok (RNS és ROS) fokozott termelődését, az eNOS szétkapcsolódását, a NO csökkent biohasznosíthatóságát és a cGMP-bontó enzim, a foszfodiészteráz (PDE)-5 megnövekedett expresszióját (1). A PDE-5-gátlás által fokozott cGMP szignalizáció kardioprotektív hatását számos szívbetegségben, ide értve a diabéteszes cardiomyopathiát, igazolták (3, 9–14).
A fenti mechanizmus mellett oxidatív stressz esetén az sGC-enzim szerkezete is károsodik, elveszíti hem prosztetikus csoportját, így NO-ra érzéketlenné válik, és inaktív állapotba kerül (15). A fentiek alapján az olyan gyógyszerek, amelyek képesek az sGC-t újra aktiválni kardioprotektív hatásúak lehetnek. Irodalmi adatok szerint az sGC aktivátor cinaciguat (BAY 58-2667) az sGC NO-ra érzéketlen formájának hemkötő zsebéhez kapcsolódik, így helyreállítja annak cGMP-termelését (15). Újabb tanulmányok a cinaciguat jótékony hatását bizonyították miokardiális infarktus (16), miokardiális iszkémiás-reperfúziós károsodás modelljeiben (17, 18), nitro-oxidatív stressz által indukált endothelialis diszfunkcióban (19), vaszkuláris neointima-képződés esetén (20) és a patológiás szívizom-hipertrófia megelőzésében (21).
A cinaciguat biztonságosságát és tolerálhatóságát humán klinikai vizsgálatokban (fázis I.) értékelték (22). Bár a cinaciguat hatékonyságát akut dekompenzált szívelégtelen betegeknél igazolták (23), a szer intravénás alkalmazása során nagy számban jelentkező nemkívánatos mellékhatás (hipotenzió) miatt (24, 25) az azonos indikációval végzett II.b fázisú vizsgálatnak idő előtt véget kellett vetni. Mindezek ellenére, a vizsgálatot végzők véleménye szerint a szer hosszú távú orális adagolás esetén kifejtheti jótékony hatásait az endothel- és a miokardiális funkció megőrzésére (26).
Fentiek alapján, jelen tanulmányunkban azt vizsgáltuk, hogy a cinaciguat hosszú távú adagolásával megőrizhető-e a bal kamrai (BK) szisztolés és diasztolés funkció, és megelőzhetőek-e a DM-hez társult miokardiális elváltozások streptozotocinnal (STZ) indukált 1-es típusú DM (T1DM) patkánymodelljében.
Módszerek
Állatok
Vizsgálatainkat a kísérleti állatok tartásáról és felhasználásáról szóló nemzetközi szabályoknak (Guide for the Care and Use of Laboratory Animals, US National Institues of Health 1996; 85–23 and EU Directive 2010/63/EU guidelines) megfelelően végeztük (etikai engedély száma: 22.1/1162/3/2010). Nyolchetes hím Sprague–Dawley patkányokat (n=46; testtömeg: 225–250 g) szabványos laboratóriumi körülmények között tartottunk.
DM-indukció
T1DM-t STZ egyszeri ip. injekcióval (60 mg/kg) váltottunk ki patkányainkban (3). Az STZ-t citrát pufferben oldottuk fel (0,1 M). A kontrollállatok csak pufferoldatot kaptak. A vércukorszintet az injekció után 72 órával, a farokvénából nyert vérből határoztuk meg. A 15 mmol/l feletti nem éhomi vércukorszinttel rendelkező állatokat tekintettük cukorbetegnek, és vontuk be a vizsgálatba.
Kísérleti csoportok, krónikus kezelési protokoll
A DM diagnózisa után a patkányokat random módon négy csoportba soroltuk: placebóval kezelt kontroll (Co; n=12), cinaciguattal kezelt kontroll (CoCin; n=12), placebóval kezelt cukorbeteg (DiabCo; n=12), és cinaciguattal kezelt cukorbeteg (DiabCin; n=10). Az állatokat 8 héten át 0,5% metilcellulóz (Co, DiabCo) vivőanyaggal, illetve cinaciguattal (CoCin, DiabCin; 10 mg/kg/nap) kezeltük p.o. Az állatok testtömegét 2 naponta mértük és a cinaciguat dózisát ehhez igazítottuk.
Hemodinamikai mérések
A BK funkciójának megítélésre BK-i nyomás-térfogat (P-V) analízist végeztünk nyomás-konduktancia mikrokatéter segítségével (MPVS-Ultra, Millar Instruments, Houston, TX, USA). Az állatokat ip. ketamin (100 mg/kg)-xylazin (3 mg/kg) injekcióval altattuk el (3, 27), majd mértük az artériás középnyomást (MAP), szívfrekvenciát (HR), a BK-i maximális szisztolés nyomást (LVSP), végdiasztolés nyomást (LVEDP), a maximális és minimális dP/dt-t (dP/dtmax dP/dtmin), a BK-i relaxáció időállandóját (Tau), az ejekciós frakciót (EF), verőmunkát (SW) és a perctérfogatot (CO). A BK-i végszisztolés P–V-összefüggés meredekségét (Ees) és az ún. „preload recruitable stroke work” (PRSW) paramétert számítottuk ki, mint a BK-i kontraktilitás érzékeny, elő- és utóterheléstől független indexeit. A BK-i diasztolés falmerevség jellemzésére a BK-i végdiasztolés P–V-összefüggés meredekségét (EDPVR) használtuk.
Biokémiai mérések
Digitális vércukormérővel, a farokvénából vett vérből vércukormérést végeztünk. A hemodinamikai méréseket követően a vena cava inferiorból gyűjtöttünk vért, amelyből plazmát különítettünk el. A plazma cGMP-szinteket enzim-immunoassay (EIA; Amersham cGMP EIA Biotrak System, GE Healthcare, Buckinghamshire, Egyesült Királyság) segítségével állapítottuk meg.
Miokardiális génexpressziós vizsgálatok
A BK génexpressziós vizsgálatára kvantitatív, valós-idejű polimeráz láncreakciót (qRT-PCR) hajtottunk végre (28). A BK-i szövetmintákat homogenizáltuk és teljes RNS-t izoláltunk. A következő géneket vizsgáltuk: pitvari nátriuretikus faktor (ANF), α-miozin nehéz lánc (MHC) és β-MHC (a β/α-MHC expressziós arányt a patológiás szívizomsejt-hipertrófia markereként használtuk), antiapoptotikus Bcl-2, proapoptotikus BAX, eNOS, kollagén 1a1 (Col1), 3a1 (Col3) és fibronektin (miokardiális fibrózis markerek), mátrix metallopeptidáz (MMP)-2 és MMP-9 és endogén inhibitoraik, MMP szöveti inhibitor (TIMP)-1 és TIMP-2 (szívizom-remodelláció markerei), különböző antioxidáns rendszerek tagjai, mint a kataláz, thioredoxin-1, glutation-reduktáz, szuperoxid-dizmutáz (SOD) és a hősokk-protein 70 (HSP70a1). A génexpressziós adatokat glicerinaldehid-3-foszfát dehidrogenázra (GADPH) normalizáltuk és egy pozitív kalibrátorra vonatkoztattuk.
Immunblot analízis
BK-i szívizomszövetet homogenizáltunk, majd a proteineket gélelektroforézisnek vetettük alá. A proteineket nitrocellulóz membránra blottoltuk és elsődleges antitestekkel inkubáltuk. A vizsgált célpontjaink a következők voltak: eNOS, sGC β1, PDE-5, PKG, vazodilatátor-stimulált foszfoprotein (VASP) és foszfo-VASP (a PKG-aktivitás markereként), profibrotikus mediátor transzformáló növekedési faktor (TGF)-β1, MMP-2 és MMP-9. A membránokat tormaperoxidázzal konjugált másodlagos antitesttel inkubáltuk. Az immunblotokat kemilumineszcens módszerrel hívtuk elő. A fehérje denzitásokat kvantifikáltuk, a denzitás értékeket GADPH-ra normalizáltuk és a kontrollcsoport átlagértékéhez viszonyítottuk.
Szövettan, immunhisztokémia
A szívizomzat fibrotikus átépülését és egyéb hisztopatológiai jellemzőit hematoxillin-eozin (HE) és Masson trikróm (MT) festett metszeteken vizsgáltuk. A szívizomsejt-hipertrófia megítélésének céljából HE-metszeteken 100 hosszanti metszetű, azonos síkból származó cardiomyocyta transznukleáris szélességét mértük (cardiomyocyta átmérő, KD). Immunhisztokémiai vizsgálatot hajtottunk végre a fibrózis marker fibronektinre, a profibrotikus mediátor TGF-β1-re, a másodlagos hírvivő cGMP-re, és a nitro-oxidatív stressz marker nitrotirozinra (NT). A szövettani és immunhisztokémiai analízist két egymástól független, a vizsgálati csoportokat nem ismerő vizsgáló végezte.
Terminal deoxi-nukleotidil-transzferáz dUTP nick end labeling (TUNEL) vizsgálat
A DNS-száltörések vizsgálatára TUNEL-festést hajtottunk végre egy kereskedelmi forgalomban kapható termékkel (DeadEnd™ Colorimetric TUNEL System, Promega, Mannheim, Germany). A TUNEL-pozitív sejtmagokat minden metszet 20 véletlenszerűen kiválasztott látóterében 2 vizsgáló számolta meg. Az adatokat a kontrollcsoport átlagértékére normalizáltuk.
Statisztika
Adatainkat átlag±SEM formában tüntettük fel. Az adatok normáleloszlásának tesztelése után kétszempontos varianciaanalízist hajtottunk végre, amelynek faktorai a diabétesz és a gyógyszeres kezelés voltak. P<0,05-ös szignifikanciaküszöbbel dolgoztunk. Tukey HSD post-hoc tesztet alkalmaztuk a csoportok közti különbségek megítélésére. Az adatokat, amelyek nem mutattak normáleloszlást, logaritmikus transzformációnak vetettük alá.
Eredmények
Testtömeg, szívtömeg és vércukorszint
Mindkét DM-csoportban szignifikánsan csökkent a szívtömeg és testtömeg, de a szívtömeg/testtömeg arány megnőtt (1. táblázat), ami a kontrollokhoz képest szignifikánsan magasabb vércukor és ivóvízfogyasztás értékekkel társult. A cinaciguat kezelés nem befolyásolta a vércukorszinteket, de csökkent ivóvízfogyasztást eredményezett.

A cinaciguat hatása a plazma és miokardiális cGMP-szintekre DM-ben
A cinaciguat kezelés a kontrollokban nem, de diabéteszes állatokban kifejezett plazma cGMP-szint-növekedéshez vezetett (1. a ábra). A cGMP-immunhisztokémia alapján a cukorbeteg állatok BK-i myocardiumának cGMP-szintje szignifikánsan alacsonyabb volt a kontrollokéhoz képest. Cinaciguat kezelés hatására ez a kontrollok szintjén maradt (1. b, c ábra).
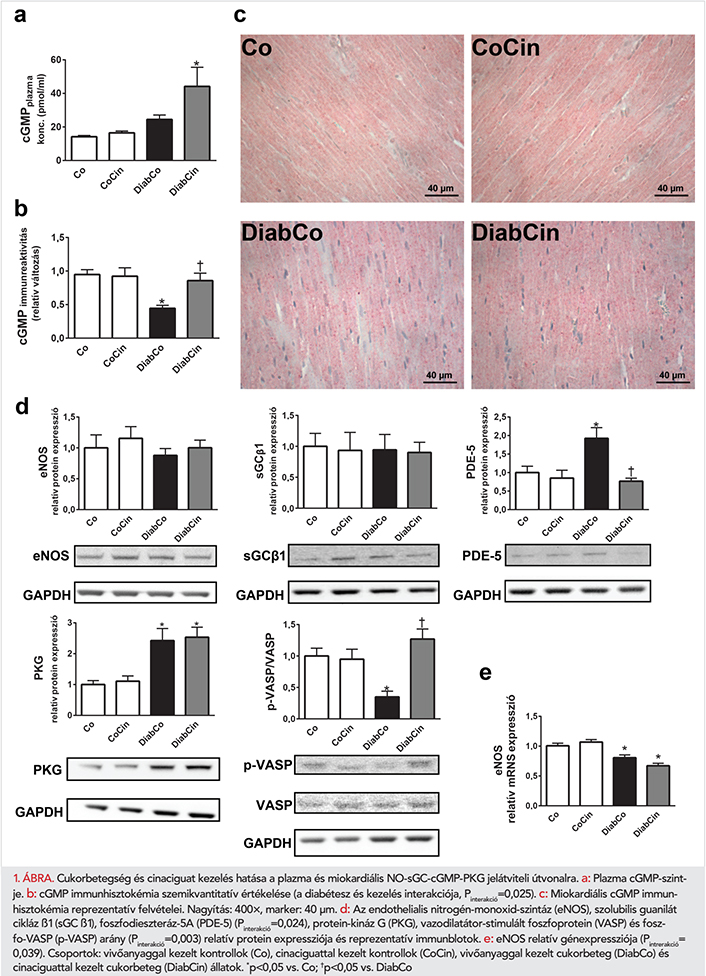
A cinaciguat hatása a NO-sGC-cGMP-PKG útvonalra
Az eNOS és sGC β1 fehérje-expressziójában nem mutatkozott különbség az egészséges és a cukorbeteg patkányok között (1. d ábra), de az eNOS génexpressziója mindkét cukorbeteg csoportban szignifikánsan alacsonyabb volt (1. e ábra). Megemelkedett PDE-5 és PKG fehérje-expressziót detektáltunk a DiabCo csoportban, habár a p-VASP/VASP arány (a PKG aktivitás markere) szignifikánsan alacsonyabb volt (a PKG aktivitásának csökkenését jelezve) (1. d ábra). A cinaciguat szignifikánsan csökkentette a PDE-5 expressziót és növelte a PKG-aktivitást (megnőtt p-VASP/VASP arány). A PKG expressziója nem mutatott különbséget a csoportok között (1. d ábra).

A DM és a cinaciguat kezelés hatása a miokardiális nitro-oxidatív stresszre
DM esetén a BK-szívizomszövetben megemelkedett NT immunreaktivitást tapasztaltunk, ami jelentős nitro-oxidatív stresszre utal. Ezt a cinaciguat kezelés szignifikáns mértékben csökkentette (2. a, b ábra). Ezek mellett a HSP70a1, kataláz, glutation-reduktáz és tioredoxin-1 fokozott expresszióját láttuk DM-ben (2. c ábra). Eközben a DiabCin csoport HSP70a1 és glutation-reduktáz mRNS-expressziója az egészséges kontrollok szintjén maradt (2. c ábra). A SOD-2 expresszióban nem mutatkozott különbség a csoportok közt (2. c ábra).
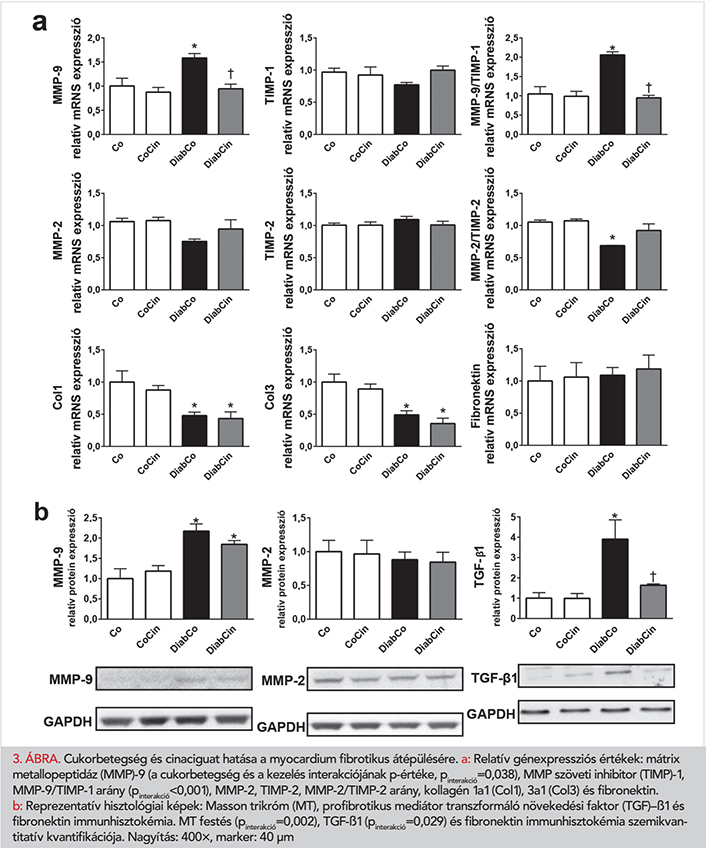
A cinaciguat hatása a miokardiális fibrózisra
DM-ben az MMP-rendszer szabályozási zavarát tapasztaltuk, amelyet megnövekedett MMP-9/TIMP-1 és lecsökkent MMP-2/TIMP-2 génexpressziós arányok jellemeztek (3. a ábra). Ezen változások a DiabCin csoportban enyhébbnek mutatkoztak. Bár a fibronektin expressziója változatlan maradt, a Col1 és Col3 expressziós szintek szignifikánsan alacsonyabbak voltak mindkét DM-csoportban (3. a ábra). A profibrotikus TGF-β1 fokozott expressziót mutatott DM-állatokban, amit a cinaciguat szignifikáns mértékben enyhített (3. b ábra). Az MMP-9 expressziója kétszeresére nőtt, míg az MMP-2 expresszió változatlan maradt DM-ben (3. b ábra). Ezekre a paraméterekre a cinaciguatnak nem volt szignifikáns hatása (3. b ábra). A myocardium MT-metszetein súlyos miokardiális fibrózist találtunk. Továbbá a profibrotikus mediátor TGF-β1 és a fibrózis marker fibronektin emelkedett immunreaktivitását láttuk cukorbeteg szívben (3. c ábra). A cinaciguat ugyanakkor csökkentette a diabéteszes szívizom MT-festésének intenzitását és a kontroll szintjén tartotta a TGF-β1 immunreaktivitást (3. c ábra).

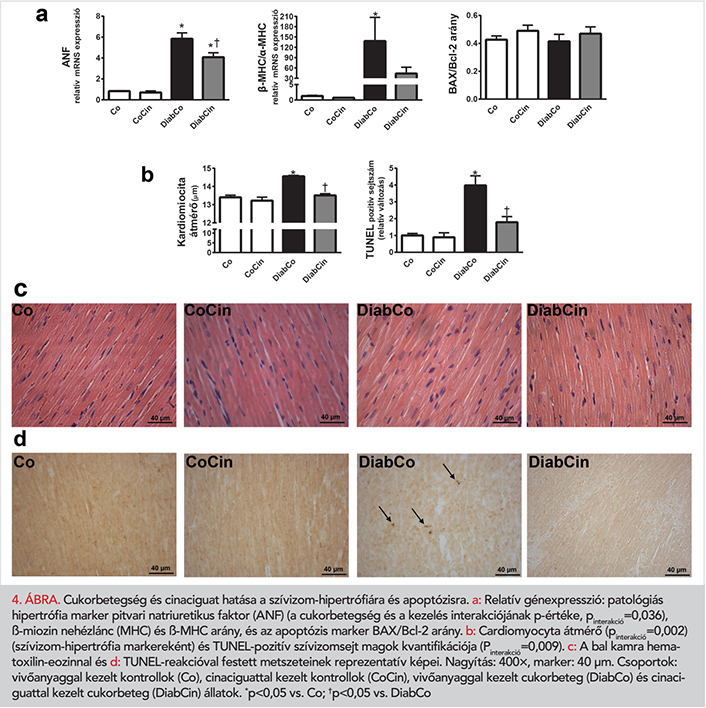
A cinaciguat hatása a szívizom-hipertrófiára és apoptózisra
A diabéteszes patkányok BK-i myocardiumából elvégzett génexpressziós vizsgálat adatai az ANF és a β-MHC/α-MHC-arány kifejezett növekedését mutatták (4. a ábra). A cinaciguat kezelés szignifikáns mértékben csökkentette az ANF expresszióját, míg a β-MHC/α-MHC-arány enyhe csökkenése volt megfigyelhető (4. a ábra). DM-ben a megnövekedett KD a szívizomsejtek hipertrófiájára utalt a DiabCo csoportban. Ezt az eltérést cinaciguat alkalmazása teljesen megelőzte (4. b, c ábra). A proapoptotikus BAX és az antiapoptotikus Bcl-2 génexpressziós szintjeiben nem volt eltérés, így a BAX/Bcl-2 arány sem változott (4. a ábra). Diabéteszben megnőtt a BK-i myocardiumban a TUNEL-pozitív sejtmagok száma, ami fokozott DNS-fragmentációra utal (4. b, c ábra). A DiabCin csoportban ugyanakkor jelentősen csökkent a TUNEL-pozitivitás (4. b, c ábra).
A cinaciguat javította a pumpafunkciót diabéteszben
A DiabCo csoportban csökkent MAP, LVSP, EF, SW, dP/dtmax és csökkent dP/dtmin értékeket láttunk, míg az LVEDP és a Tau megnőtt. Ezen adatok a BK szisztolés és diasztolés diszfunkciójára utaltak. A HR szignifikánsan csökkent a DM-csoportokban, de CO nem különbözött jelentős mértékben a csoportok között. A MAP, LVSP, SW, dP/dtmax és dP/dtmin változatlan maradt a DiabCin csoportban, de az LVEDP és a Tau szignifikánsan javult cinaciguat hatására cukorbetegségben. Emellett a kezelés hatására az EF javuló tendenciát mutatott (p=0,054) (2. táblázat). A kontraktilitás-indexek (Ees, PRSW) értékei szignifikánsan csökkentek cukorbeteg állatokban, súlyos szisztolés funkciózavarra utalva (5. a, b ábra). A gyógyszeres kezelés viszont a PRSW szignifikáns növekedéséhez vezetett, míg az Ees javuló tendenciát mutatott cukorbetegségben (p=0,092) (5. b ábra). A Co csoporttal összehasonlítva az EDPVR meredeksége növekvő tendenciát mutatott (p=0,063) DM-állatokban, amit a cinaciguat kezelés szignifikáns mértékben csökkentett (5. b ábra). A cinaciguatnak nem volt hemodinamikai hatása a kontrollokban.


Megbeszélés
Tanulmányunkban az sGC aktivátor cinaciguat diabéteszes cardiomyopathiára kifejtett hatását vizsgáltuk 1-es típusú cukorbetegség patkánymodelljében. Kimutattuk, hogy a cinaciguat hosszú távú adagolásával hatékonyan emelhető mind a plazma, mind pedig a myocardium cGMP-szintje és helyreállítható a cGMP-PKG-jelátvitel aktivitása a szívizomban. Ezek a molekuláris változások a szívizom-hipertrófia csökkenéséhez valamint a szisztolés és diasztolés funkció javulásához vezettek állatmodellünkben.
Az STZ által indukált T1DM-patkánymodellt széles körben alkalmazzák a diabéteszes cardiomyopathia vizsgálatára és új kezelési lehetőségeinek kutatására. Diabétesz indukciója során az STZ a glükóz-transzporter 2-n keresztül kerül a hasnyálmirigy Langerhans béta-sejtjeibe, így biztosítva annak szelektív toxicitását. Jelen vizsgálatunkban eredményes STZ-indukciót követően, az állatok emelkedett vércukorszintje és megnőtt napi ivóvízfogyasztása bizonyította a diabétesz kialakulását (29).
Cukorbetegségben a kialakuló szövődmények elsődleges oka feltehetően a magas vércukorszint. A hyperglykaemia számos mechanizmuson és jelátviteli útvonalon keresztül képes különböző kardiovaszkuláris betegségeket előidézni. Annak ellenére, hogy több tanulmány is vizsgálta a diabéteszes cardiomyopathia patomechanizmusát, a pontos kórélettani elváltozások mindmáig nem ismertek. Többek között a magas vércukorszint növeli a ROS- és RNS-termelést, következményes nitro-oxidatív stressz kialakulását és az antioxidáns rendszerek és hősokk proteinek expressziójának növekedését előidézve (1, 30). Vizsgálatunkban bizonyítottuk a jelenlévő fokozott nitro-oxidatív stresszt (NT-pozitivitás) (31) a myocardiumban, ami hozzájárulhatott az endogén antioxidáns rendszerek indukciójához és a jelentős hősokk-válaszhoz. Számos negatív hatásuk mellett a reaktív szabad gyökök az sGC-enzim oxidációját okozzák, ami így elveszti hem prosztetikus csoportját és NO-érzéketlen, inaktív állapotba kerül. A NO-cGMP-jelátvitel további károsodásához a PDE-k különböző típusai között fellépő egyensúlyzavar is hozzájárulhat (32). A csökkent intracelluláris cGMP szintek miatt az NO-cGMP-PKG-jelátvitel inaktiválódik a diabéteszes szívben. Ezzel összhangban a cukorbeteg patkányainkban csökkent a miokardiális cGMP-szintje, míg az sGC β1-protein expressziója nem változott. Ezeken felül cukorbetegségben csökkent az NO-cGMP-PKG útvonal aktivitása, amit az eNOS csökkent génexpressziója, emelkedett PDE-5 fehérjeszintek (korábbi adatokkal összhangban) (33) és a PKG-aktivitást jelző VASP szignifikánsan csökkent foszforilációja (alacsony p-VASP/VASP arány) (21) jeleztek. Mindazonáltal emelkedett PKG protein-expressziós szinteket figyeltünk meg, ami a BK egy kevéssé hatékony kompenzációs mechanizmusaként értelmezhető. A diabéteszes állatokban tapasztalható négyszer nagyobb mértékű DNS-károsodásban a cGMP-jelátvitelhez köthető citoprotektív folyamatok károsodása játszhatott szerepet. Érdekes módon, diabéteszben a csökkent miokardiális cGMP-koncentráció ellenére a plazma cGMP-szint változatlan maradt. A jelenség hátterében valószínűsíthető a cukorbetegségben tapasztalt ANF-emelkedés és ennek következményeként az egyéb szervekben fokozódott nátriuretikus peptid és partikuláris guanilát-cikláz (pCG) aktiváció kompenzáló hatása. Így a megtartott plazma cGMP-szint forrásának az egyéb szövetekből kiszivárgó intracelluláris cGMP tekinthető (34).
A cinaciguat betegségspecifikus hatását (csak az sGC oxidált, inaktivált formáját képes aktiválni) (15), számos fokozott nitro-oxidatív stresszel járó patológiás állapotban bizonyították (18, 19). Ezzel összefüggésben cinaciguat alkalmazásával jelen vizsgálatunkban jelentős citoprotektív hatást értünk el cukorbetegségben. Ezt bizonyítja többek között a csökkent NT-festődés (nitro-oxidatív stressz jele), az endogén antioxidáns rendszerek és hősokk-proteinek normalizálódott expressziója, és a DNS-törések csökkent mértéke. A mechanizmus, amelyen keresztül a szer csökkenti az oxidatív stresszt ugyanakkor még további vizsgálatok tárgya. Egy lehetséges magyarázat, hogy a fokozott cGMP-jelátvitel által a különböző NADPH-oxidázok aktivitása csökken (35). A csökkent oxidatív terhelés következtében a PDE-5-enzim termelődése visszatért a kontrollok szintjére, ami hozzájárulhatott az intracelluláris/plazma cGMP-szint növekedéséhez. A fokozott cGMP-termelés fokozta a PKG-jelátvitel (emelkedett p-VASP/VASP arány) aktivitását cukorbetegségben.
A diabéteszes cardiomyopathia és a szívizomzat kóros átépülése hátterében több folyamat együttesen áll úgy, mint a szív hipertrófiája, fibrózisa és a szöveti sérülést követő DNS-károsodás és apoptózis (1). Ezzel összefüggésben jelen vizsgálatunkban, cukorbetegségben a szívizomzat patológiás hipertrófiáját találtuk, amelynek jelei a megnövekedett KD, fokozódott ANF-expresszió és a β-/α-MHC arány eltolódása volt. Az sGC aktivációjának antihipertrófiás hatásai nemcsak szövettani (csökkent KD), hanem molekuláris szinten (csökkent ANF-expresszió, tendencia a β-/α-MHC arány csökkenésére) is jelentkeztek. Eredményeink összecsengenek korábbi irodalmi adatokkal (21), amelyben igazolták a cinaciguat hipertrófiát és fibrózist gátló hatását sejtkultúra kísérletekben. A diabéteszes szívizom fibrotikus átépülésében jelentős szerepet játszik az MMP-k szabályozási zavara (1), a fokozott fibroblaszt-proliferáció (36), a csökkent Col1 és Col3 mRNS-expresszió (37) és a megnövekedett TGF-β1 útvonal (38). A fentiekkel összhangban jelen vizsgálatunkban a TGF-β1 profibrotikus mediátor fokozott termelődését és a Col1 és Col3 mRNS-szintjének csökkenését, intenzív MT és fibronektin festődést és fokozott DNS-fragmentációt találtunk diabéteszben. Az sGC-enzim gyógyszeres aktivációja ugyanakkor hatékonyan gátolta a szív patológiás elváltozásainak kialakulását az MMP szabályozási zavar helyreállításával, a TGF-β1 szint csökkentésével, a fibrotikus változások megelőzésével és a szívizom TUNEL-pozitivitásának csökkentésével. Érdekesség, hogy csoportjaink között a BAX/Bcl-2 arányban nem volt különbség, ami arra enged következtetni, hogy a klasszikus apoptotikus útvonalaknak közvetlenül nincs szerepe a megfigyelt hatásokban. A fentiek magyarázataként az sGC-cGMP jelátvitel és a TGF-β szignalizáció kapcsolata szolgálhat (39).
Korábbi irodalmi adatok szerint diabéteszben a szív szisztolés és diasztolés funkciója jelentősen károsodik (2, 29). Ezzel összhangban jelen vizsgálatunkban, cukorbetegségben szignifikáns kardiális diszfunkciót találtunk. A MAP szignifikáns csökkenése (valószínűleg a diabéteszes polyuria következményeként) (40) mellett, a DiabCo csoport szignifikánsan romlott pumpafunkciót (csökkent LVSP, SW, EF és dP/dtmax) mutatott. Az EF és a dP/dtmax széles körben alkalmazott kontraktilitás paraméter, ugyanakkor ezek jelentősen függenek a szív elő- és utóterhelésétől, illetve a szívfrekvenciától (27). A fentiek kiküszöbölésére P–V-analízist végeztünk, aminek során elő- és utóterheléstől független, érzékeny kontraktilitás paramétereket számoltunk (Ees, PRSW) (27). A BK-i kontraktilitás paraméterei súlyos szisztolés funkciózavart mutattak a DiabCo csoportban. Az sGC-aktiváció eredményeként a szisztolés funkció figyelemre méltó javulását értük el diabéteszes állatokban: szignifikánsan emelkedett a PRSW, ami jelzi a BK-i kontraktilitás javulását, míg az EF és az SW az egészséges kontrollok szintjére emelkedett. A diasztolés diszfunkciót szignifikánsan károsodott BK-i aktív relaxáció (csökkent dP/dtmin, megnyúlt Tau) és jelzetten emelkedett BK-i falmerevség (magasabb LVEDP, tendenciózusan megnőtt EDPVR) jellemezte cukorbetegségben. Ezzel szemben, cinaciguat nemcsak az aktív relaxációt (csökkent Tau), de a diasztolés falmerevséget is (csökkent LVEDP és EDPVR) javította.
Tanulmányunk az első, amely az sGC farmakológiai aktivációjának hatását vizsgálta diabéteszes cardiomyopathiában. Vizsgálatunk eredményeiből megállapítható, hogy az sGC-aktivátor cinaciguat a vércukorszintre kifejtett hatás nélkül javítja a diabéteszes cardiomyopathia kórtani jellemzőit. A cinaciguat jótékony hatásainak molekuláris mechanizmusai közé tartozik a NO-sGC-cGMP-PKG tengely aktivációja, illetve különböző antioxidáns, antihipertrófiás és antifibrotikus jelátviteli folyamatokra gyakorolt előnyös hatása diabéteszben. Mindezek alapján az sGC farmakológiai aktivációja potenciális terápiás megközelítés lehet a cukorbetegség kardiovaszkuláris szövődményeinek kezelésére.
Jelen közlemény Mátyás et al. Cardiovasc Diabetol 2015; 14: 145. közleményének (41) felhasználásával készült a BioMed Central engedélye (Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/) alapján.
Köszönetnyilvánítás
Jelen munkánkat a Magyar Tudományos Akadémia Bolyai János Kutatási Ösztöndíja (R.T.), az Országos Tudományos Kutatási Alapprogramok PD100245 programja (R.T.), az Emberi Erőforrás Támogatáskezelő (Nemzeti Tehetség Program; NTP-NFTÖ-16-0081; M.Cs.) és a Nemzeti Kutatási, Fejlesztési és Innovációs Hivatal – NKFIH (NVKP-16-1-2016-0017) támogatta. Elismerés illeti Biró Henriett, Fritz Gábor, Alt Gábor, Fischinger Tímea, Fazekas Krisztina és Gregor Viktória munkáját. Köszönet illeti Johannes-Peter Stascht (Bayer Healthcare, Wuppertal, Németország), amiért a munkához a cinaciguatot biztosította.
Irodalom
1. Huynh K, Bernardo BC, McMullen JR, et al. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther 2014; 375–415. DOI: https://doi.org/10.1016/j.pharmthera.2014.01.003
2. Radovits T, Korkmaz S, Loganathan S, et al. Comparative investigation of the left ventricular pressure-volume relationship in rat models of type 1 and type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 2009; H1: 25–33. DOI: https://doi.org/10.1152/ajpheart.00165.2009
3. Radovits T, Bomicke T, Kokeny G, et al. The phosphodiesterase-5 inhibitor vardenafil improves cardiovascular dysfunction in experimental diabetes mellitus. Br J Pharmacol 2009; 909–19. DOI: https://doi.org/10.1111/j.1476-5381.2008.00098.x
4. Joffe, II, Travers KE, Perreault-Micale CL, et al. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. J Am Coll Cardiol 1999; 2111–9. DOI: https://doi.org/10.1016/S0735-1097(99)00436-2
5. Pearson JT, Jenkins MJ, Edgley AJ, et al. Acute Rho-kinase inhibition improves coronary dysfunction in vivo, in the early diabetic microcirculation. Cardiovasc Diabetol 2013; 111. DOI: https://doi.org/10.1186/1475-2840-12-111
6. Cai L. Suppression of nitrative damage by metallothionein in diabetic heart contributes to the prevention of cardiomyopathy. Free Radic Biol Med 2006; 851–61. DOI: https://doi.org/10.1016/j.freeradbiomed.2006.06.007
7. Sharma V, McNeill JH. Diabetic cardiomyopathy: where are we 40 years later? Can J Cardiol 2006; 305–8. DOI: https://doi.org/10.1016/S0828-282X(06)70914-X
8. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 1991; 109–42.
9. Radovits T, Arif R, Bomicke T, et al. Vascular dysfunction induced by hypochlorite is improved by the selective phosphodiesterase-5-inhibitor vardenafil. Eur J Pharmacol 2013; 110–9. DOI: https://doi.org/10.1016/j.ejphar.2013.04.012
10. Kukreja RC, Salloum FN, Das A. Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. J Am Coll Cardiol 2012; 1921–7. DOI: https://doi.org/10.1016/j.jacc.2011.09.086.
11. Szabo G, Radovits T, Veres G, et al. Vardenafil protects against myocardial and endothelial injuries after cardiopulmonary bypass. Eur J Cardiothorac Surg 2009; 657–64. DOI: https://doi.org/10.1016/j.ejcts.2009.03.065
12. Hirschberg K, Radovits T, Loganathan S, et al. Selective phosphodiesterase-5 inhibition reduces neointimal hyperplasia in rat carotid arteries after surgical endarterectomy. J Thorac Cardiovasc Surg 2009; 1508–14. DOI: https://doi.org/10.1016/j.jtcvs.2008.10.016
13. Loganathan S, Radovits T, Hirschberg K, et al. Effects of selective phosphodiesterase-5-inhibition on myocardial contractility and reperfusion injury after heart transplantation. Transplantation 2008; 1414–8. DOI: https://doi.org/10.1097/TP.0b013e31818aa34e
14. Schafer A, Fraccarollo D, Pfortsch S, et al. Improvement of vascular function by acute and chronic treatment with the PDE-5 inhibitor sildenafil in experimental diabetes mellitus. Br J Pharmacol 2008; 886–93. DOI: https://doi.org/10.1038/sj.bjp.0707459
15. Stasch JP, Schmidt PM, Nedvetsky PI, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 2006; 2552–61. DOI: https://doi.org/10.1172/JCI28371
16. Korkmaz S, Radovits T, Barnucz E, et al. Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation 2009; 677–86. DOI: https://doi.org/10.1161/CIRCULATIONAHA.109.870774
17. Salloum FN, Das A, Samidurai A, et al. Cinaciguat, a novel activator of soluble guanylate cyclase, protects against ischemia/reperfusion injury: role of hydrogen sulfide. Am J Physiol Heart Circ Physiol 2012; H1347–54. DOI: https://doi.org/10.1152/ajpheart.00544.2011
18. Radovits T, Korkmaz S, Miesel-Groschel C, et al. Pre-conditioning with the soluble guanylate cyclase activator Cinaciguat reduces ischaemia-reperfusion injury after cardiopulmonary bypass. Eur J Cardiothorac Surg 2011; 248–55. DOI: https://doi.org/10.1016/j.ejcts.2010.05.025
19. Korkmaz S, Loganathan S, Mikles B, et al. Nitric oxide- and heme-independent activation of soluble guanylate cyclase attenuates peroxynitrite-induced endothelial dysfunction in rat aorta. J Cardiovasc Pharmacol Ther 2013; 70–7. DOI: https://doi.org/10.1177/1074248412455696
20. Hirschberg K, Tarcea V, Pali S, et al. Cinaciguat prevents neointima formation after arterial injury by decreasing vascular smooth muscle cell migration and proliferation. Int J Cardiol 2013; 470–7. DOI: https://doi.org/10.1016/j.ijcard.2012.01.032
21. Irvine JC, Ganthavee V, Love JE, et al. The soluble guanylyl cyclase activator bay 58–2667 selectively limits cardiomyocyte hypertrophy. PLoS One 2012; e44481. DOI: https://doi.org/10.1371/journal.pone.0044481
22. Frey R, Muck W, Unger S, et al. Pharmacokinetics, pharmacodynamics, tolerability, and safety of the soluble guanylate cyclase activator cinaciguat (BAY 58–2667) in healthy male volunteers. J Clin Pharmacol 2008; 1400–1. DOI: https://doi.org/10.1177/0091270008322906
23. Lapp H, Mitrovic V, Franz N, et al. Cinaciguat (BAY 58–2667) improves cardiopulmonary hemodynamics in patients with acute decompensated heart failure. Circulation 2009; 2781–8. DOI: https://doi.org/10.1161/CIRCULATIONAHA.108.800292
24. Levy PD, Laribi S, Mebazaa A. Vasodilators in Acute Heart Failure: Review of the Latest Studies. Curr Emerg Hosp Med Rep 2014; 126–32. DOI: https://doi.org/10.1007/s40138-014-0040-z
25. Erdmann E, Semigran MJ, Nieminen MS, et al. Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur Heart J 2013; 57–67. DOI: https://doi.org/10.1093/eurheartj/ehs196
26. Gheorghiade M, Greene SJ, Filippatos G, et al. Cinaciguat, a soluble guanylate cyclase activator: results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur J Heart Fail 2012; 1056–66. DOI: https://doi.org/10.1093/eurjhf/hfs093
27. Pacher P, Nagayama T, Mukhopadhyay P, et al. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 2008; 1422–34. DOI: https://doi.org/10.1038/nprot.2008.138
28. Radovits T, Olah A, Lux A, et al. Rat model of exercise-induced cardiac hypertrophy: hemodynamic characterization using left ventricular pressure-volume analysis. Am J Physiol Heart Circ Physiol 2013; H1: 24–34. DOI: https://doi.org/10.1152/ajpheart.00108.2013
29. Litwin SE, Raya TE, Anderson PG, et al. Abnormal cardiac function in the streptozotocin-diabetic rat. Changes in active and passive properties of the left ventricle. J Clin Invest 1990; 481–8. DOI: https://doi.org/10.1172/JCI114734
30. Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, et al. Heat shock proteins and cardiovascular pathophysiology. Physiol Rev 2001; 1461–97.
31. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007; 315–424. DOI: https://doi.org/10.1152/physrev.00029.2006
32. Lukowski R, Krieg T, Rybalkin SD, et al. Turning on cGMP-dependent pathways to treat cardiac dysfunctions: boom, bust, and beyond. Trends Pharmacol Sci 2014; 404–13. DOI: https://doi.org/10.1016/j.tips.2014.05.003
33. Fang L, Radovits T, Szabo G, et al. Selective phosphodiesterase-5 (PDE-5) inhibitor vardenafil ameliorates renal damage in type 1 diabetic rats by restoring cyclic 3’,5’ guanosine monophosphate (cGMP) level in podocytes. Nephrol Dial Transplant 2013; 1751–61. DOI: https://doi.org/10.1093/ndt/gfs391
34. Hamet P, Pang SC, Tremblay J. Atrial natriuretic factor-induced egression of cyclic guanosine 3’:5’-monophosphate in cultured vascular smooth muscle and endothelial cells. J Biol Chem 1989; 12364–9.
35. Koka S, Das A, Salloum FN, et al. Phosphodiesterase-5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med 2013; 80–8. DOI: https://doi.org/10.1016/j.freeradbiomed.2013.01.031
36. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014; 660–71. DOI: https://doi.org/10.1007/s00125-014-3171-6
37. Van Linthout S, Seeland U, Riad A, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 2008; 319–27. DOI: https://doi.org/10.1007/s00395-008-0715-2
38. Cai L, Li W, Wang G, et al. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002; 1938–48. DOI: https://doi.org/10.2337/diabetes.51.6.1938
39. Beyer C, Zenzmaier C, Palumbo-Zerr K, et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFbeta signalling. Ann Rheum Dis 2015; 1408–16. DOI: https://doi.org/10.1136/annrheumdis-2013-204508
40. Wang S, Mitu GM, Hirschberg R. Osmotic polyuria: an overlooked mechanism in diabetic nephropathy. Nephrol Dial Transplant 2008; 2167–72. DOI: https://doi.org/10.1093/ndt/gfn115
41. Matyas C, Nemeth BT, Olah A, et al. The soluble guanylate cyclase activator cinaciguat prevents cardiac dysfunction in a rat model of type-1 diabetes mellitus. Cardiovasc Diabetol 2015; 145. DOI: https://doi.org/10.1186/s12933-015-0309-x