Identification of a mitochondrial gene mutation in a systemic disease manifesting primarily as hypertrophic cardiomyopathy
█ Case report
DOI: 10.26430/CHUNGARICA.2017.47.2.135
Authors:
Tringer Annamária1, Grosz Zoltán2, Nagy Viktória1, Gál Anikó2, Csányi Beáta1, Lidia Hategan1, Borbás János1, Gavallér Henriette1, Pálinkás Eszter1, Forster Tamás1, Molnár Mária Judit2, Sepp Róbert1
1Szegedi Tudományegyetem, II. sz. Belgyógyászati Klinika és Kardiológiai Központ, Szeged
2Semmelweis Egyetem, Genomikai Medicina és Ritka Betegségek Intézete, Budapest
Summary
Mitochondrial diseases belong to a heterogeneous group of multisystem diseases as rare systemic disorders caused by mutations of the mitochondrial genome (mtDNA) and cc. 1500 nuclear genes which are responsible for the mitochondrial function. The disease primarily affects organs with high energy expenditure as the central nervous system and skeletal muscle, but numerous other forms are known, in which disease-specific symptoms may be present. The disease typically shows maternal inheritance. In our study genetic analysis was performed in a patient presenting with the cardiac phenotype of hypertrophic cardiomyopathy.
A 32-years-old female patient, with short stature (140 cm), came to medical attention at the age of 27 when she presented with exertional dyspnea and chest discomfort. Hearing disorder was known for years, which was attributed to bilateral cochlear lesion. ECG showed short PQ interval and signs of left ventricular hypertrophy. Echocardiographic and MRI examinations confirmed non-obstructive hypertrophic cardiomyopathy, enlarged left atrium, severe concentric left (LVmax: 15 mm) and right ventricular hypertrophy (RVmax: 8 mm). During the course of the disease type I diabetes mellitus developed and then visual disturbances appeared, due to a confirmed retinal dystrophy. Her laboratory findings showed elevated LDH, CK, troponin T, and NTproBNP values. Neurological status was negative. Her brother was known to have stroke, epilepsy, septal hypertrophy, hyperhomocysteinaemia, and he died at age of 17 due to recurrent strokes. Her mother has been known for having hearing disorder and diabetes mellitus.
Genetic screening for sarcomere gene mutations and Fabry disease causing mutations for the GLA gene was negative. Analysis of the mitochondrial genome confirmed a m.3243A>G mutation, most commonly found in MELAS (mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes) syndrome.
Our case points out to a possible mitochondrial disease and the need for genetic testing in cases of hypertrophic cardiomyopathy with systemic involvement.
ISSUE: CARDIOLOGIA HUNGARICA | 2017 | VOLUME 47, ISSUE 2
Összefoglalás
A mitokondriális betegségek több szervrendszert érintő, a mitokondriális genom (mtDNS) és a mitokondrium működéséhez szükséges kb. 1500 gén mutációi által okozott ritka szisztémás betegségek. A betegség elsősorban a nagy energiaigényű szervek, mint központi idegrendszer és a vázizom betegségeinek képében jelentkezik, de számos más formája ismert, amelyeknél a tünetek betegség specifikusan jelennek meg. Az mtDNS-hibák következtében kialakuló betegségek típusosan anyai öröklésmenetet mutatnak.
Munkánkban egy hipertrófiás cardiomyopathia kardiális fenotípusát mutató beteg genetikai analízisét végeztük el. Az észlelésekor 32 éves, alacsony termetű (140 cm) nőbeteg kardiális panaszai 27 éves korában kezdődtek, terhelésre jelentkező nehézlégzés, mellkasi diszkomfort formájában. Évek óta ismert hallászavara hátterében kétoldali cochlearis léziót igazoltak. EKG-ján rövid PQ-távolság és balkamra-hipertrófia volt látható. Echokardiográfiás és MR-vizsgálata nonobstruktív hipertrófiás cardiomyopathiát igazolt, tágabb bal pitvarral, kifejezett koncentrikus bal (LVmax: 15 mm) és jobbkamra-hipertrófiával (RVmax: 8 mm). Észlelése során inzulinterápiát igénylő diabetes mellitus alakult ki, majd látászavar jelentkezett, amelynek hátterében retina-disztrófiát véleményeztek. Laborjában magasabb LDH, CK, troponin T- és NTproBNP-értékeket észleltünk. Neurológiai státusza negatív volt. Fiútestvérénél stroke, epilepszia, septum-hipertrófia, hyperhomocystinaemia volt ismert, 17 éves korában recidív stroke-ok következtében halálozott el. Édesanyjánál diabetes mellitus és hallászavar szerepel az anamnézisben.
Genetikai vizsgálata szarkomer génmutációk és Fabry-kórt okozó GLA-mutációk irányában negatív volt. A mitokondriális genom vizsgálata az m.3243A>G szubsztitúciót találta, amelyet leggyakrabban korábban a MELAS (mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes) szindrómával asszociáltan írtak le.
Esetünk a hipertrófiás cardiomyopathia képével járó, de szisztémás érintettséget mutató kórképek esetén mitokondriális betegség lehetőségére és ez irányú genetikai vizsgálat szükségességére hívja fel a figyelmet.
Bevezetés
A hipertrófiás cardiomyopathia (HCM) a myocardium primer megbetegedése, amelynek legfontosabb morfológiai jellemzője a kifejezett szívizom-hipertrófia (1). A HCM típusos esetben a szarkomer fehérjéit kódoló mutációk következtében alakul ki, de az esetek 5-10%-ában egyéb, specifikus géneltérések állhatnak a betegség hátterében, amelyek utánozzák a HCM morfológiai és klinikai megjelenését, ilyenkor HCM fenokópiákról beszélünk (Fabry-betegség, Danon-betegség, transthyretin amyloidosis stb.). A HCM-fenokópiák öröklődés módja, klinikai lefolyása és kezelése eltér a szarkomer géneket érintő mutációk által kialakuló HCM-es betegekétől, ezért utóbbiak elkülönítése a szarkomer-mutációk által okozott HCM-től nagy jelentőségű, mert adott esetben a diagnózis pontosításán túl specifikus terápiás implikációi lehetnek (2).
Esetismertetésünkben egy dominálóan hipertrófiás cardiomyopathia morfológiai képében jelentkező, de szisztémás érintettség jeleit is mutató beteg esetét mutatjuk be, akiben az mtDNS m.3243A>G szubsztitúcióját azonosítottunk a betegség hátterében.
Esetismertetés
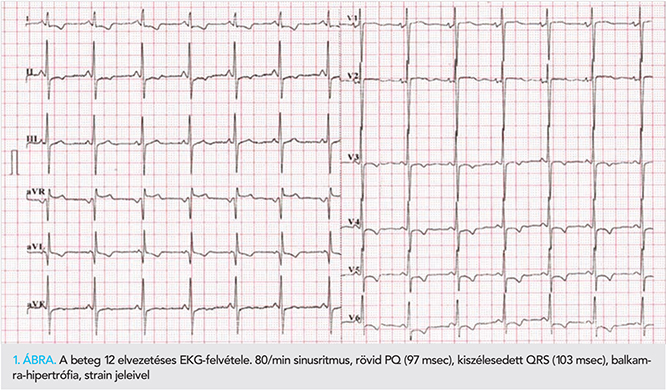
Az klinikai észlelésekor 32 éves, kifejezetten alacsony termetű (140 cm) nőbeteg kardiális panaszai 27 éves korában kezdődtek, terhelésre jelentkező nehézlégzés, mellkasi diszkomfort formájában. Évek óta ismert hallászavara hátterében kétoldali cochlearis léziót igazoltak. EKG-ján rövid, 97 ms-os PQ-távolság és balkamra-hipertrófia volt látható, strain jeleivel (1. ábra). Echokardiográfiás vizsgálata nonobstruktív hipertrófiás cardiomyopathiát igazolt, tágabb bal pitvarral, kifejezett koncentrikus bal és jobbkamra-hipertrófiával. A koncentrikus balkamra-hipertrófia a bal kamra valamennyi szegmentumát és a papilláris izmokat is érintette, 15 mm-es maximális bal kamrafal vastagsággal az anterior septum középső szegmentumában (2. ábra). A globális BK-funkció megtartott volt, szegmentális falmozgászavar nem volt észlelhető. Kifejezett, restriktív típusú diasztolés diszfunkció jeleit észleltük (E/A: 86/35; DCT: 186 ms, Ea: 6 cm/s, E/Ea: 14,3). Szív MR-vizsgálata az echokardiográfiás lelettel egyező morfológiai képet mutatott (3. ábra), késői típusú kontraszthalmozással a laterális fal középső (11-12-es szegmentum), a posterior fal középső (10-es szegmentum), inferior fal basalis (4-es szegmentum) szegmentumaiban és az anterior falon (1-7-13-as szegmentumok) (3. C és D ábra). Laborjában magasabb LDH (463–539 U/l), CK (170–193 U/l), troponin T (0,046–0,082 ng/ml) és NTproBNP (644–1179 pg/ml) értékeket észleltünk. Vese- és májfunkciós értékei, vérképe lényegi kórosat nem mutatott, kiemelendő, hogy laktátszintje is normális volt (1,3–2,0 mmol/l). Kiterjesztett endokronológiai laborparaméterei hasonlóképpen kóros eltérést nem mutattak. A beteg észlelése során inzulinterápiát igénylő diabetes mellitus alakult ki, majd látászavar jelentkezett, amelynek hátterében retina-disztrófiát véleményeztek. Neurológiai státusza negatív volt. Családi anamnéziséből kiemelendő, hogy fiútestvérénél alacsony testalkat, stroke, epilepszia, septum-hipertrófia, hallászavar volt ismert, 17 éves korában recidív stroke-ok következtében halálozott el. Édesanyjánál diabetes mellitus és hallászavar volt ismert, kardiológiai vizsgálata lényegi organikus kardiális eltérést nem igazolt.

A szarkomer fehérjéit kódoló gének új generációs szekvenálásával 7 szarkomer génvariánst észleltünk, amelyek közül egyik sem volt egyértelműen patogén. Mindössze a béta-myozin nehéz lánc gént (MYH7) érintő p.Asp1450Asn variánsról van publikált adat, amelyet nem HCM-es, hanem egy DCM-es betegcsoportban azonosítottak. Utóbbiak alapján fenti génvariánsok patogén szerepét a betegség kialakulásában nem lehet egyértelműen állítani, esetleg a fenotípus kialakulásában modifikátorként közreműködhettek. Genetikai vizsgálata Fabry-kórt okozó GLA-mutációk irányában is negatív volt.

A mitokondriális genom vizsgálata viszont a 3243. nukleoidnál egy A/G-pontmutációt igazolt (m.3243A>G), 38%-os heteroplazmia arányban. Ezt a mitokondriális DNS-mutációt leggyakrabban a MELAS-szindrómával (mitokondriális encephalomyopathia, laktát-acidózis és stroke-like epizódok) asszociáltan írták le.
Fentiek alapján esetünket dominálóan HCM képében megjelenő mitokondriális DNS-betegségnek tartjuk.
Megbeszélés
A primer mitokondriális betegségek több szervrendszert érintő ritka multiszisztémás betegségek, amelyeket a mitokondrium optimális működéséért felelős gének: a mitokondriális genom (mtDNS) és kb. 1500 nukleáris gén hibája okoz. A kórkép bármely szervet érintheti, jelentős morbiditással jár (3). Az mtDNS-t érintő betegségek prevalenciája 1:5000-hez, és 1:200-hoz az esélye, hogy egy újszülött potenciálisan patogén mtDNS-mutáció hordozó legyen (4). Az egyes szövetek eltérő mennyiségben tartalmaznak mitokondriumokat, energiaigényüktől függően. Az mtDNS mutációi elsősorban azokat a szerveket károsítják, amelyek működése különösen energiaigényes. Ennek megfelelően elsősorban az idegrendszert (görcsrohamok, ataxia, dementia, encephalopathia formájában), a vázizomzatot (gyengeség, fáradékonyság, myopathia formájában) és a szívet (cardiomyopathia, vezetési zavar formájában) érinti (5) de számos más megjelenési formája ismert, ilyen az alacsony termet, szenzorineuronális halláscsökkenés, epilepsziás rohamok, ophthalmoplegia externa, fejfájás, rekurráló stroke, myopathia, cardiomyopathia, encephalopathia, ataxia, neuropathia, demencia, ciklikus hányás, terhelési intolerancia (6). A betegség első tünetei különböző életkorban jelentkezhetnek. Típusos esetben a stroke-szerű neurológiai gócjelek már a második évtizedben megjelenhetnek.
Az mtDNS-betegségben szenvedő egyén sejtjeiben a vad típusú és mutáns mtDNS-molekulák különböző arányban, együtt vannak jelen; mely jelenséget heteroplazmiának hívnak. Utóbbi magyarázza a szervspecifikus manifesztációkat, amelynek súlyossága nem mindig korrelál a heteroplazmia arányával (4).
A mitokondriális DNS-betegségekre maternális öröklődés jellemző, ennek oka, hogy csak a petesejt mitokondriumai kerülnek az embrióba. Így csak az anya örökítheti tovább a betegséget, ha az anya mtDNS-betegségben szenved az utódoknak nagy az esélye, hogy örökölik a betegséget. Az mtDNS-betegségben szenvedő hölgyek egyes petesejtjeinek 0 és 100% között változhat a heteroplazmia aránya, így ezekben az esetekben nem tudjuk megbecsülni a következő generációban a betegség ismétlődésének kockázatát (7).
A mitokondriális DNS zárt, cirkuláris, kettősláncú DNS, amelyet 16 569 bázis alkot, szekvenciája 37 gént kódol. Az mtDNS rendkívül gazdaságos, nincsenek benne intronok és csak minimális nem kódoló DNS-szakaszt tartalmaz. A legjellemzőbb génvariáns, az m.3243A>G-variáns, a MELAS-szindrómás esetek több mint 80%-áért felelős (8). Az m.3243A>G-variáns egy pontmutáció, ami az MT-TL1-gént érinti, és a mitokondriális transzfer RNS egyik leucinját károsítja [tRNALeu(UUR)]. Ezzel a mitokondriális transzlációs folyamat megszakad, amely a légzési lánc károsodásához, és az aerob metabolizmusban csökkent ATP-termelődéshez vezet. Bár a MELAS-betegek többségükben az m.3243A>G variánst hordozzák, ez a mutáció nem specifikus MELAS-szindrómára, hiszen más betegségekben úm. progresszív ophtalmoplegia externában (PEO), cardiomyopathiában, sensorineurális süketségben és anyai öröklődést mutató diabetes mellitusban is megtalálható (3). Bár a m.3243A>G-mutációhordozó betegek klinikai képét általában a neurológiai tünetek dominálják, fenti betegek nagy többségében a normális echokardiográfiás leletek ellenére szív MRI-vizsgálattal korán megjelenő abnormis kardiális funkció mutatható ki, amelynek foka jól korrelál a vázizomban megtalálható mutáns mtDNS arányával (9, 10).
A mitokondriális betegségekben észlelt cardiomyopathia típusos esetben hipertrófiás cardiomyopathia, de dilatatív cardiomyopathia is lehet. Hipertrófiás cardiomypathia esetén diagnosztikus „red flag”-ek, vészjelzők hívhatják fel figyelmünket mitokondriális cardiomyopathia lehetőségére. Utóbbiak közül a tanulási nehézség, mentális retardáció, hallászavar, látászavar, izomgyengeség, ptosis, bal kamra falmozgászavara, rövid PQ-intervallum, AV-blokk emelendők ki (1, 2).
A mitokondriális betegségeknek jelenleg effektív oki kezelése nincsen, a terápia jórészt a tünetek kezelésére szorítkozik, és érdemben nem befolyásolja a betegség kimenetelét. A diagnózis felállítása után fontos a szervi manifesztációk kezelése, úm. a diabétesz kezelése, ritmuszavarok kontrollálása, szükség esetén pacemaker-beültetés, hallászavar esetén cochlearis implantáció, görcsrohamok esetén antiepileptikumok használata, katarakta kezelése, ptosis korrekciója. Laktát-acidózis esetén javasolt a glükózbevitel korlátozása. Izomgyengeséggel küzdő betegeknek ajánlott a moderált edzés. Számos nem specifikus gyógyszeres kezelés lehetőségét vetették fel az utóbbi években, amelyek hatékonyságára eddig nincs meggyőző eredmény. Tapasztalati úton tiamin, riboflavin, Q10 (400 mg/nap), L-karnitin (3×1000 mg), C-vitamin és magas zsírtartalmú diéta javasolt. MELAS-szindróma esetén kerülendő gyógyszerek a tetraciklinek, amiodaron, barbiturát, kloramfenikol, valproát, carboplatin, zidovudin, interferonok, β-blokkolók, acetilszalicilsav, statinok, amelyek súlyosbíthatják a tüneteket a légzési lánc gátlásán keresztül. Bár a többszervi érintettséget mutató betegségek a szívtranszplantáció relatív kontraindikációját jelentik, szívtranszplatáció szóba jöhet abban az esetben, ha a klinikai megjelenés a szívre korlátozódik és más szervek érintettsége enyhe, és nem progresszív. Specifikus, mitokondriális szinten ható szerek klinikai vizsgálata folyamatban van (pl. a mitokondriális permeabilitást befolyásoló pórus inhibitor bendavia vagy az antioxidáns ciszteamin-bitartrát). A betegség prognózisa nagyban függ az első tünetek megjelenésének idejétől. Minél korábbi életkorban jelennek meg a tünetek, a betegség lefolyása annál progresszívebb.
Esetünk dominálóan hipertrófiás cardiomyopathia képével járó, de szisztémás érintettséget mutató kórképek esetén mitokondriális betegség lehetőségére hívja fel a figyelmet és hangsúlyozza az ez irányú genetikai vizsgálat szükségességét.
Köszönetnyilvánítás
A munka a „Nemzeti Agykutatási Program” és a „Ritka betegségek patogenezisének kutatása, új diagnosztikai és terápiás eljárásokat megalapozó fejlesztések” program (GINOP-2.3.2-15-2016-00039) segítségével készült.
Irodalom
1. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733–79.
https://doi.org/10.1093/eurheartj/ehu284
2. Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 1448–58.
https://doi.org/10.1093/eurheartj/ehs397
3. Zeviani M, Di Donato S. Mitochondrial disorders. Brain 2004; 127(10): 2153–72. doi:https://doi.org/10.1093/brain/awh259
4. Cree LM, et al. The inheritance of pathogenic mitochondrial DNA muatarions. Biochim Biophys Acta 2009; 1792: 1097–1102.
https://doi.org/10.1016/j.bbadis.2009.03.002
5. Hsu YHR, Yogasundaram H, Parajuli N, et al. MELAS Syndrome and Cardiomyopathy: Linking Mitochondrial Function to Heart Failure Pathogenesis. Heart Fail Rev 2016; 21: 103–116.
https://doi.org/10.1007/s10741-015-9524-5
6. Sunde K, Blackburn PR, Cheema A, et al. Case report: 5 year follow-up of adult late-onset mitochondrial encephalomyopathy with lactic acid and stroke-like episodes (MELAS). Mol Genet Metab Rep 2016; 9: 94–97.
https://doi.org/10.1016/j.ymgmr.2016.11.002
7. Dean NL, Battersby BJ, Ao A, Gosden RG, Tan ST, Shoubridge EA, Molnar MJ. Prospects of preimplantation genetic diagnosis for heritable mitochondrial DNA diseases. Mol Hum Reprod 2003; 9: 631–638.
https://doi.org/10.1093/molehr/gag077
8. Lorenzoni PJ, Werneck LC, Kay CSK, et al. When should MELAS (Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like episodes) be the diagnosis? Arq Neuropsiquiatr 2015; 73(11): 959–967.
https://doi.org/10.1590/0004-282X20150154
9. Kieren G, Hollingsworth A, Grainne S, et al. Cardiomyopathy is common in patients with the mitochondrial DNA m.3243A>G mutation and correlates with mutation load. Neuromuscular Disorders 2012; 22: 592–596.
https://doi.org/10.1016/j.nmd.2012.03.001
10. Remenyi V, Inczedy-Farkas G, Komlosi K, et al. Retrospective assessment of the most common mitochondrial DNA mutations in a large Hungarian cohort of suspect mitochondrial cases. Mitochondrial DNA: PART A 2015; 26(4): 572–578.
https://doi.org/10.3109/19401736.2013.878901