Reverse electrical remodeling following pressure unloading in a rat model of myocardial hypertrophy
█ Original article
DOI: 10.26430/CHUNGARICA.2018.48.2.118
Authors:
Ruppert Mihály1,2*, Barta Bálint András2*, Korkmaz-Icöz Sevil1, Li Shiliang1, Oláh Attila2, Mátyás Csaba2, Németh Balázs Tamás2, Benke Kálmán2, Sayour Alex Ali2, Karck Matthias1, Merkely Béla2, Radovits Tamás2*, Szabó Gábor1*
1Ruprecht-Karls Egyetem Szívsebészeti Klinika, Kísérleti Kutató Laboratórium, Heidelberg
2Semmelweis Egyetem, Kísérleti Kutató Laboratórium, Városmajori Szív- és Érsebészeti Klinika, Budapest
*A szerzők azonos mértékben járultak hozzá a közleményhez
Készült Ruppert et al. Hypertens Res 2017; 40: 637–645. közleményének felhasználásával a SpringerNature Kiadó írásos engedélyével (1)
Summary
Introduction: Pressure overload-induced left ventricular myocardial hypertrophy (LVH) is characterized by increased proarrhythmic vulnerability. In contrast, pressure unloading leads to reverse remodeling and decreases LVH-associated arrhythmogenicity. However, cellular changes that occur during reverse electrical remodeling have been studied less. Therefore, we aimed to provide an electrocardiographic characterization of a rat model of LVH that underwent pressure unloading and to simultaneously identify the underlying cellular and functional alterations.
Methods: LVH was induced in rats by abdominal aortic banding for 6 or 12 weeks. Sham-operated animals served as controls. Pressure unloading was evoked by removing the aortic constriction after week 6 (Debanded). Serial echocardiography and electrocardiography were performed to investigate the development and the regression of LVH. Protein expression levels were detected by western blot. Myocardial fibrosis was assessed by Picrosirius red staining.
Results: Pressure unloading resulted in the regression of LVH in correlation with the reversion of the prolonged corrected QT interval (cQT: 68.7±1.6 vs. 91.0±1.9 ms Debanded vs. AB week 12, P<0.05). Furthermore, pressure unloading prevented the functional decompensation of LVH and simultaneously preserved adequate atrioventricular conduction (PQ: 47.5±1.2 vs. 53.8±1.9 ms Debanded vs. AB week 12, P<0.05). Finally, pressure unloading effectively preceded the broadening of the QRS complex (QRS: 21.8±0.5 vs. 24.9±0.7 ms Debanded vs. AB week 12, P<0.05) in parallel with the attenuation of interstitial collagen accumulation.
Conclusions: The regression of LVH with maintained cardiac function and decreased myocardial fibrosis contributes to pressure unloading-induced reverse electrical remodeling.
ISSUE: CARDIOLOGIA HUNGARICA | 2018 | VOLUME 48, ISSUE 2
Összefoglalás
Bevezetés: Széles körben elfogadott, hogy a fokozott nyomásterhelés által előidézett bal kamrai (BK) szívizom-hipertrófia (BKH) kialakulását a myocardium fokozott aritmia hajlama kíséri. Szintén ismert, hogy a fokozott utóterhelés megszüntetése az átépült szívizomszerkezet visszaalakulását, ún. reverz remodellációját eredményezheti, amely együtt járhat az aritmogenitás egyidejű csökkenésével. Ennek ellenére a myocardium reverz elektromos átépülését kísérő sejtszintű változásokat kevesen vizsgálták. Ezért jelen tanulmányunkban célunk volt a BKH kialakulásának és visszafejlődésének elektrokardiográfiás nyomonkövetése, és az elektrofiziológiai változások hátterében meghúzódó sejtszintű és funkcionális történések azonosítása.
Módszerek: Patkánymodellünkben a fokozott nyomásterhelést az aorta műtéti beszűkítésével (aortic banding; AB) biztosítottuk 6 (AB-6hét), illetve 12 hét (AB-12hét) időtartamra. A szívizom-hipertrófia kialakulása után, a reverz remodellációt a szűkület eltávolításával (Debanding) idéztük elő. A kontrollcsoportot áloperált állatok alkották. A BKH kialakulását, és visszafejlődését elektrokardiográfiával és szívultrahang-vizsgálattal követtük. A fehérje-expressziót western blottal tanulmányoztuk. A miokardiális fibrózist picrosirius vörös festéssel vizsgáltuk.
Eredmények: A szűkület eltávolítása a BKH jelentős visszafejlődését eredményezte, amelynek mértéke korrelált a megnyúlt korrigált QT-intervallum regressziójával. (cQT: 68,7±1,6 vs. 91,0±1,9 ms Debanded vs. AB-12hét, p<0,05). A fokozott nyomásterhelés megszüntetése továbbá eredményesen előzte meg a bal kamra funkcionális hanyatlását, és egyúttal megőrizte a pitvar-kamrai átvezetést (PQ: 47,5±1,2 vs. 53,8±1,9 ms Debanded vs. AB-12hét, p<0,05). Mindemellett az utóterhelés csökkentése megakadályozta a QRS-komplexum kiszélesedését (QRS: 21,8±0,5 vs. 24,9±0,7 ms Debanded vs. AB-12hét, p<0,05), amellyel az intersticiális kollagén mennyiségének csökkenése állt párhuzamban.
Következtetések: A BKH visszafejlődése a szívfunkció romlásának és a miokardiális fibrózis súlyosbodásának gátlásával együtt hozzájárul a nyomáscsökkenés által kiváltott reverz elektromos remodellációhoz.
Bevezetés
A bal kamra hosszan tartó nyomásterhelésének (pl. hipertónia, aortastenosis) hatására bal kamrai szívizom-hipertrófia fejlődik ki (BKH), amely a malignus kamrai aritmiák és így a hirtelen szívhalál alapvető rizikófaktora (2). Ennek magyarázatául szolgál, hogy a krónikusan fennálló hemodinamikai terhelés nemcsak a szívizomsejtek méretének növekedésével, hanem jellegzetes elektrofiziológiai változásokkal is együtt jár. Erre utal az elektromos remodelláció fogalma, amely fokozott intersticiális fibrózist (3), a Ca2+ homeosztázis fehérjéinek megváltozott expresszióját (4) valamint a réskapcsolatok lecsökkent számát foglalja magába (5). Ezen celluláris változások a BKH kialakulása során eltérő időben jelentkeznek, és eltérő mechanizmussal vezetnek kamrai aritmiákhoz.
A BKH-t kísérő leggyakoribb elektrofiziológiai eltérés az akciós potenciál (AP) megnyúlása, amely az EKG-n megnyúlt QT-intervallum formájában jelenik meg. Hátterében repolarizációs zavar áll (3), amely a csökkent kifelé irányuló K+ (6), illetve a fokozott befelé irányuló Ca2+-áramra (4) vezethető vissza. A repolarizációs idő megnyúlása a korai és a késői utódepolarizációkra egyaránt hajlamosít (7, 8). Ezzel összhangban a korán, funkcionális szempontból még kompenzált stádiumban lévő BKH mellett fellépő hirtelen szívhalál esetekért elsősorban triggerelt aktivitások felelnek (3).
Továbbá a vezetés lassulására utaló QRS-komplexum kiszélesedése szintén gyakran megfigyelhető dekompenzált BKH-ban és szívelégtelenségben (9), amit a jelentős miokardiális fibrózis (10), és a réskapcsolatok alacsony száma magyarázhat (5). A vezetés sebessége viszont nem egyenlő mértékben lassul minden szívizomterületen, ezért a myocardium refrakteritás szempontjából inhomogénné válik (11), amely folyamatok együttesen re-entry típusú kamrai tachyarrhythmiák kialakulására hajlamosítanak.
Továbbá a BKH-t és szívelégtelenséget kísérő elektrofiziológiai változások a pitvarokban is manifesztálódnak. Ezt támasztja alá, hogy szívelégtelen nyulakban szignifikánsan hosszabb PR-intervallumot, a pitvar-kamrai junkció megnyúlását és bizonyos ioncsatorna expressziós eltéréseket találtak (12).
A fokozott nyomásterhelés megszüntetése ugyanakkor, megfelelően korai stádiumban, az átépült szívizomszerkezet visszaalakulását, ún. reverz remodellációját eredményezheti (13, 14), amely előnyös elektrofiziológiai változásokkal is együtt járhat. Utóbbit támasztották alá Rials és munkatársai (15), akik aorta szűkített majd a szűkületet eltávolított macskamodellben a BKH regresszióját követően normalizálódott monofázisos AP hosszról (APH), szignifikánsan magasabb fibrillációs küszöbről és kevesebb kiváltható polimorf kamrai aritmiáról számoltak be. Mindezek ellenére jelenleg nem áll rendelkezésre adat a reverz elektromos remodelláció mechanizmusairól.
Ezért patkánykísérletünkben célunk volt (1) a BKH kialakulásának és visszafejlődésének elektrokardiográfiával történő karakterizációja, valamint (2) a reverz elektromos remodelláció háttérében meghúzódó sejtszintű és funkcionális elváltozások egyidejű azonosítása.
Módszerek
Állatok
Hím Sprague–Dawley patkányokat (n=51) (160–180 g; Charles River, Sulzfeld, Németország) szabványos laboratóriumi körülmények között tartottunk. Vizsgálatainkat a kísérleti állatok tartásáról és felhasználásáról szóló nemzetközi szabályoknak (Guide for the Care and Use of Laboratory Animals, US National Institues of Health 1996; 85–23) megfelelően végeztük, az etikai bizottság engedélyével.
A hasi aorta beszűkítésére és a szűkület eltávolítására alkalmazott eljárások
A bal kamra fokozott nyomásterhelését a hasi aorta műtéti beszűkítésével idéztük elő (aortic banding, AB) (n=33), ahogy azt már korábban ismertettük (16, 17). Röviden összefoglalva, medián laparotomiát követően egy 2-0-s sebészi fonál felhasználásával az aorta abdominális szakaszát, a suprarenalis síkban egy 22 G-s tű külső átmérőjével megegyező szélességűre szűkítettük (18). Az áloperált (Sham) állatok (n=18) azonos eljáráson mentek keresztül, az aorta beszűkítésétől eltekintve. A 6. hét elteltével az aortaszűkített állatok egy részénél (n=11) a fonál eltávolításra került (Debanded), a fokozott nyomásterhelés megszüntetése céljából. A posztoperatív fájdalomcsillapítást szubkután adott buprenorfin injekcióval biztosítottuk.
Kísérleti csoportok
6 héttel az első műtétet követően állatainkat az alábbi öt csoportba osztottuk:
- 6 hetes aortaszűkített csoport (AB-6hét; n=8), a csoport tagjait aortaszűkítést követően 6 hétig követtük;
- 6 hetes áloperált csoport (Sham-6 hét; n=8), a csoport tagjait, áloperációt követően 6 hétig követtük;
- 12 hetes aortaszűkített csoport (AB-12hét; n=14), a csoport tagjait aortaszűkítést követően 12 hétig követtük;
- 12 hetes áloperált csoport (Sham-12 hét; n=10), a csoport tagjait, áloperációt követően 12 hétig követtük;
- 12 hetes aortaszűkületet eltávolított csoport (Debanded; n=11), a csoport tagjai aortaszűkítésen mentek keresztül, majd a 6. hét után a szűkület eltávolításra került és az állatokat a 12. hét végéig követtük.
A szívek elektromos, morfológiai és hemodinamikai változásait a 3., 6., 9., és 12. héten végzett elektrokardiográfiás és echokardiográfiás mérésekkel követtük nyomon. A kísérleti periódus végén invazív vérnyomásmérést hajtottunk végre, majd meghatároztuk a szív- (HW) és tüdő (LW) tömegeket és a tibia hosszát (TL).
Echokardiográfia
A vizsgálatokat a korábbiakban ismertetettek szerint hajtottuk végre (19). Röviden: az állatokat 100%-os oxigénnel történő lélegeztetés mellett 5%-os indukciós, majd 1,5-2%-os fenntartó izoflurán anesztéziának vetettük alá, miközben testhőmérsékletüket automata fűtőpad segítségével 37 °C-on tartottuk. Egy HDI 5000 CV UH-géphez (ATL Ultrasound, Philips, Bothell, WA, USA) kapcsolt 10 MHz-es lineáris fejjel két dimenziós parasternalis hosszú és rövid, valamint a papilláris izmot középen metsző M-módú felvételeket készítettünk, majd ezeket szoftveres kiértékelésnek vetettük alá (HDI Lab, Philips, Bothell, WA, USA).
A következő paramétereket határoztuk meg: BK elülső falvastagság (AWT), BK hátulsó falvastagság (PWT) diasztoléban (d) és szisztoléban (s), valamint BK végdiasztolés- (LVEDD) és végszisztolés (LVESV belső átmérő. Minden mérést háromszor hajtottunk végre, eredményeiket átlagoltuk.
A BK-i szívizomtömeget (LV mass) a módosított Devereux-módszer (20) alapján számítottuk: LVmass (g)={[(LVEDD+AWTd+PWTd)]3–LVEDD3) × 1,04} × 0,8+0,14. A végdiasztolés (LVEDV) és végszisztolés (LVESV) térfogatokat a prolate modell szerint határoztuk meg: LVEDV=[(3,14/6) × LVEDD2] × L (21). A frakcionális rövidülést (FS) és az ejekciós frakciót (EF) a következő képletek alapján számítottuk: FS=[(LVEDD–LVESD)/LVEDD] × 100 és EF=(LVEDV–LVESV/LVEDV) × 100.
Elektrokardiográfia
Szubkután elhelyezett tűelektródákkal 12 elvezetéses EKG-t regisztráltunk (Mortara Instrument, Milwaukee, WI, USA) (19). Az érzékenységet 10 mm/mV-ra, a futási sebességet 50 mm/s-ra állítottuk. A kiértékelést a II-es elvezetés alapján végeztük, amely során a következő paraméterek kerültek meghatározásra: szívfrekvencia; PQ- és QT-intervallumok, illetve QRS-komplexum hossza. QT-intervallumot a QRS első kitérésétől a T-hullám végéig mértük, és a korrekcióját (QTc) a Bazett-formula patkányokhoz igazított változatával számoltuk: [nQTc=QT/(RR/f)1/2] (22).
Invazív hemodinamikai vizsgálat
A kísérleti periódus végén invazív hemodinamikai vizsgálatot hajtottunk végre (16) pentobarbitállal (60 mg/kg, ip.) altatott, tracheotomizált és intubált patkányokon, amelyek testhőmérsékletét fűtőpad segítségével végig 37 °C-on tartottuk. A bal véna jugularis externát folyadék adagolás céljából polietilén katéterrel kanüláltuk, míg a jobb arteria carotis communison egy 2F-s nyomás- és térfogatmérő konduktancia mikrokatétert (SPR-838, Millar Instruments, Houston, Tx, USA) vezettünk az aorta ascendensbe, majd a stabilizációs idő leteltével a bal kamrába. A következő paramétereket határoztuk meg: szisztolés és diasztolés vérnyomás (sBP és dBP), artériás középnyomás (MAP) és BK-i végszisztolés (LVESP) és végdiasztolés (LVEDP) nyomásértékek. A hemodinamikai mérések végeztével az állatokat kivéreztetéssel eutanizáltuk.
Szövettan
A BK-i myocardium darabjait 4%-os paraformaldehid oldatban fixáltuk és paraffinba ágyaztuk. A hipertrófia értékelése céljából a BK 5 μm-es, transmuralis, azonos magasságból származó, hematoxillin-eozinnal festett metszetein lemértük és átlagoltuk 100 db longitudinálisan orientált cardiomyocyta transnuclearis átmérőjét (CD). A miokardiális fibrózis mértékét picrosirius vörössel festett metszeteken szemikvantitatív pontozás segítségével osztályoztuk (0: nincs jelen, 1: enyhe, 2: közepes és 3: súlyos) állatonként 20 véletlenszerűen kiválasztott látótérben.
Immunoblot analízis
Western blotot végeztünk a korábban ismertetett módon (23). BK-i szívizomszövetet homogenizáltunk, majd a fehérje oldatokat gélelektroforézisnek vetettük alá. A fehérjéket nitrocellulóz membránra blottoltuk és elsődleges antitestekkel inkubáltuk. A vizsgált célpontjaink a következők voltak: Connexin-43 (Cx43) L-típusú Ca2+-csatorna Protein α1C (LTCC) és szarkoplazmás/endoplazmás retikulum Ca2+-ATPáz 2 (SERCA2). A membránokat tormaperoxidázzal konjugált másodlagos antitesttel inkubáltuk. Az immunblotokat kemilumineszcens módszerrel (PerkinElmer, Rodgau-Juegesheim; Németország) hívtuk elő és az intenzitásokat a Chemi-smartTM 5100 rendszerrel (Peqlab, Biotechnologie, GmbH, Erlangen, Németország) detektáltuk. A fehérje denzitásokat kvantifikáltuk, a denzitás értékeket GADPH-ra normalizáltuk és tüntettük fel.
Statisztika
Minden adatot átlagérték±SEM formájában adtunk meg. Az adatok normáleloszlását Shapiro–Wilk-teszttel ellenőriztük. Normáleloszlás esetén, egy utas varianciaanalízist (ANOVA) és Tukey-féle post hoc tesztet hajtottunk végre. Normáleloszlás hiányában Kruskal–Wallis és Dunn-tesztet végeztünk. A sorozatos EKG- és UH-vizsgálatok eredményeit csoporton belül ismétléses egy utas ANOVA-val és Tukey-féle post hoc teszttel vizsgáltuk. A különböző paraméterek közti korrelációkat Spearman-teszttel igazoltuk. A p<0,05 értékeket tekintettük statisztikailag szignifikánsnak.
Eredmények
Az aorta szűkítésének és a szűkület eltávolításának hatása az artériás vérnyomásra
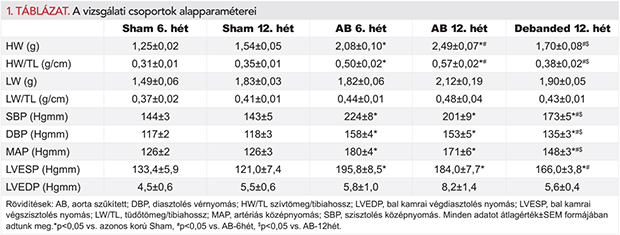
Az aorta abdominalis műtéti beszűkítése szignifikánsan emelkedett BPs-t, BPd-t és MAP-t eredményezett az AB-6hét és AB-12hét csoportokban, míg a szűkület eltávolítása az említett paraméterek csökkenését idézte elő a Debanded állatoknál (1. táblázat).
Miokardiális remodelláció és a nyomásterhelés megszüntetése által indukált reverz remodelláció
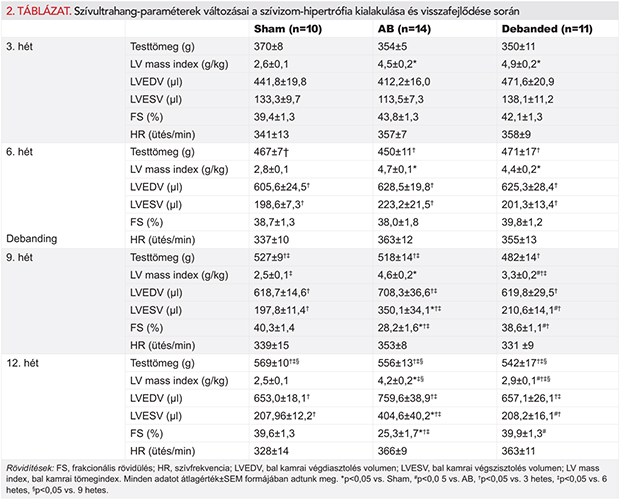
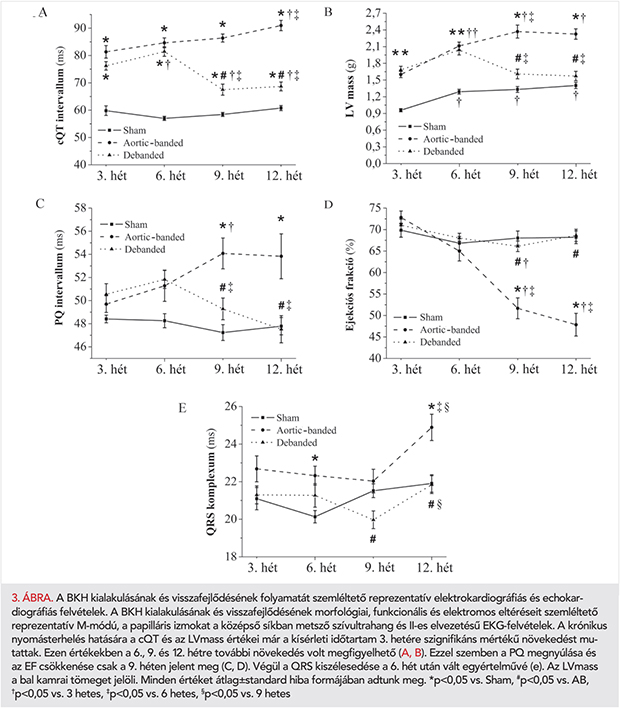
Az AB-csoportokban a BKH fennállását az ultrahanggal mért BK-i tömegindexek már a 3. hét leteltével igazolták (2. táblázat). Ezt a 6. héten megfigyelt CD és HW/TL arányok is alátámasztották (1. táblázat; 1. A, B és 3B ábrák). Ellenben a picrosirius-festett metszeteken nem találtunk különbséget a miokardiális fibrózist illetően a 6 hetes aortaszűkített csoportban (1. A és C ábrák). A 6. hét után a szívizom-hipertrófiára funkcionális szempontból kompenzált stádium volt jellemző, amelyet a megtartott EF- és FS-értékek igazoltak (2. táblázat, 1. A és 1. C ábrák).
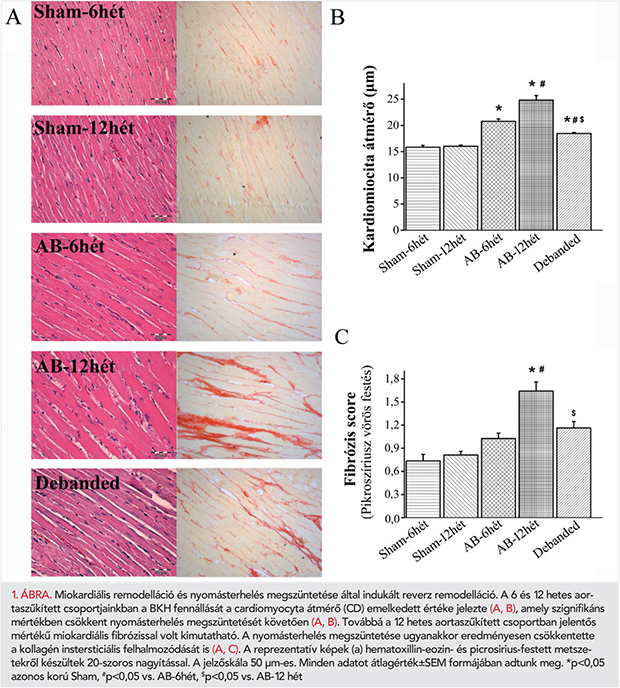
A szívfunkció romlása a 9. héten kezdődött, és ez a 12. hétre további romlást mutatott. A kísérleti periódus végéhez közelítve az AB-12hét csoportban az EF és a FS szignifikáns csökkenését, valamint LVESV jelentős növekedését mutattuk ki az AB-6hét csoporthoz viszonyítva (2. táblázat és 3. D ábra). Emellett a 12 hetes aortaszűkített csoportban az LVmass, CD, HW/TL arány és a miokardiális fibrózis szignifikáns emelkedését észleltük, akár a kontrollcsoport akár az AB-6hét csoport értékeihez hasonlítva (1. táblázat, 1. A–C ábrák). Továbbá a LVEDP és a LW/TL arány értékeiben egy erős növekvő tendencia volt megfigyelhető az AB-12hét csoportban. A nyomásterhelés megszüntetése a Debanded csoportban az AB-12hét csoporthoz képest szignifikáns mértékben csökkentette az LVmass, CD, HW/TL arány értékeit (1. táblázat; 1.A, B és 3. B ábrák) és védett az intersticiális kollagén felhalmozódással szemben is (1. A és C ábrák). A szűkület eltávolítása továbbá megelőzte a szívfunkció fokozatos hanyatlását (2. táblázat és 3. D ábra).
Az elektromos remodelláció és reverz remodelláció elektrokardiográfiás karakterizációja
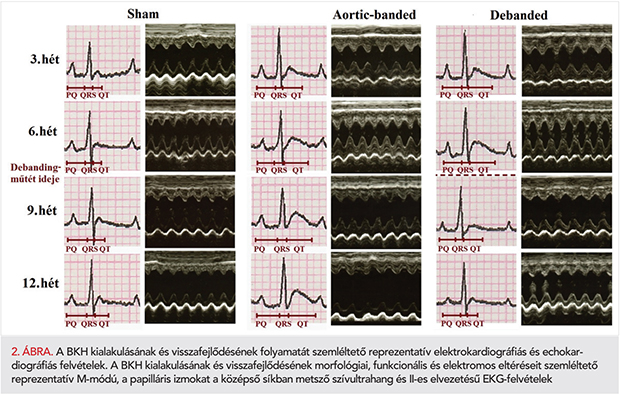
Az aortaszűkített állatoknál már a harmadik hét leteltével a QT-intervallum szignifikáns mértékű megnyúlását tapasztaltuk, ami a kísérleti periódus végéig tovább fokozódott (2. és 3. A ábrák). A QT-intervallum megnyúlása erős korrelációt mutatott az LVmass (4. A ábra), a CD (r=0,6332; p=0,0001) és a HW/TL arány értékeivel (r=0,8864; p=0,0001). A PQ-intervallum megnyúlása a 9. héttől vált megfigyelhetővé és mértéke szignifikáns összefüggést mutatott az EF (4. B ábra), a FS (r=−0,2874; p=0,0006), és az LW/TL arány értékeivel (r=0,2889; p=0,0441).
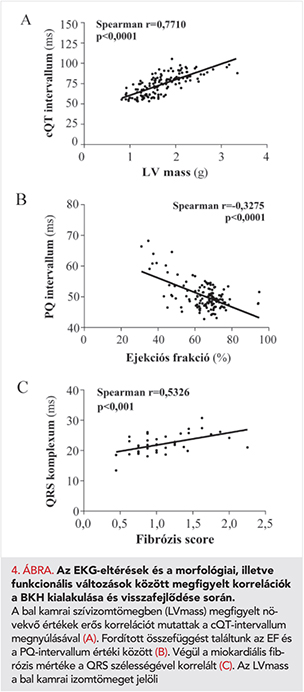
A QRS-komplexum kiszélesedését mind az AB-6hét és AB-12hét csoportnál tapasztaltuk (2. és 3. E ábra), ami jól korrelált a miokardiális fibrózis mértékével (4. C ábra), azonban a Cx43 expressziójával nem (Spearman r=−0,04027; p=0,8327). A nyomásterhelés megszüntetése az említett paraméterek (QT, PQ, QRS) szempontjából jelentős védő hatást biztosított (2–3. ábrák).
Az elektromos remodelláció és reverz remodelláció hátterében megfigyelhető fehérje expressziós változások
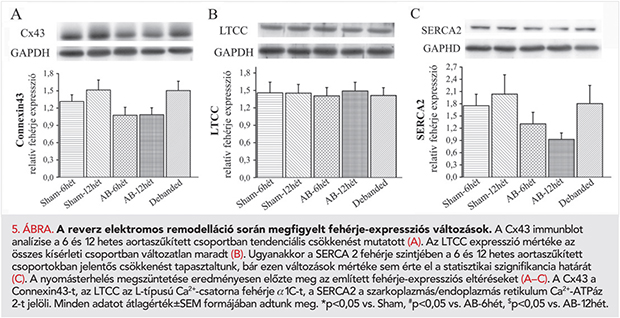
A western blot vizsgálat erős csökkenő tendenciát mutatott a Cx43 fehérje expressziójában mind az AB-6hét mind az AB-12hét csoportokban. A Debanded csoportban a Cx43 fehérje miokardiális szintje a kontrollcsoportok értékeihez tért vissza (5. A ábra). Az LTCC expressziója változatlan maradt minden kísérleti csoportban (5. B ábra). Ezzel szemben a hosszan fennálló nyomásterhelés következtében a 6 és 12 hetes aortaszűkített csoportokban tendenciálisan csökkent SERCA2-expresszió volt kimutatható, mely fehérje expressziója ugyanakkor a Debanded-csoportban a kontroll állatokra jellemző értékhez közelített (5. C ábra).
Megbeszélés
Jelen patkánykísérletünkben célunk volt a BKH kialakulásának és visszafejlődésének elektrokardiográfiával történő karakterizációja, valamint a reverz elektromos remodelláció hátterében meghúzódó elváltozások egyidejű azonosítása. Tudomásunk szerint ez az első vizsgálat, amely e kórállapotban az EKG-n tapasztalható eltérések és a sejtszintű-, illetve funkcionális változások közötti kapcsolatot vizsgálja.
A nyomásterhelés megszüntetése a BKH visszafejlődésével párhuzamosan helyreállította a megnyúlt QT intervallumot
A BKH-t kísérő leggyakoribb elektrofiziológiai eltérés az AP megnyúlása (4). Az AP időtartamának növekedése az EKG-n megnyúlt QT-intervallum formájában manifesztálódik, amelynek megjelenése a korábbi kísérletes eredmények alapján szoros korrelációt mutat a BKH kialakulásával (24, 25). Kísérletünkben a fokozott nyomásterhelés hatására a műtétet követően már a 3. héten jelentős mértékű BKH volt kimutatható, amely együtt járt a cQT-intervallum megnyúlásával (2., 3., 4. A ábra). A kialakult szívizom-hipertrófia a 6. a 9. és a 12. hétre további progresszió mutatott, amelyet a szignifikánsan növekedett LVmass, HW/TL és CD-értékei igazoltak (1. táblázat; 1. A, B, 3. B ábrák). A BKH súlyosbodása a QT-intervallum további hosszabbodását vonta maga után (2., 3.A ábra).
A BKH-ban megfigyelhető QT-intervallum megnyúlás hátterében a kifelé irányuló K+-áramok csökkenése és a befelé irányuló Ca2+-áram növekedése által előidézett meghosszabbodott repolarizációs idő állhat (4, 6). A kórélettan az AP-hosszát a repolarizációs periódus hosszától teszi függővé, amely a két antagonista ionáram egymáshoz viszonyított állapota által meghatározott. A kifelé irányuló K+-áram ugyanis a szarkolemma gyors repolarizációjához vezet, amelyet a feszültségfüggő Ca2+-csatornák nyílása azonnal ellensúlyoz (26). Az így létrejövő egyensúlyi állapotoknak végül a K+-csatornák javára bekövetkező eltolódás vet véget, amely következtében a repolarizáció teljessé válik. Ezekből következik, hogy a két említett ionáram precíz szabályozása szükséges a fiziológiás QT-intervallum fenntartásához.
A Ca2+-csatornákban bekövetkező fehérje-expressziós változásokat illetően BKH és szívelégtelenség fennállása esetén megosztottak a tudományos vélemények. A nehézséget az okozza, hogy a BKH súlyossági fokától függően egyaránt jelentették az LTCC expressziójának növekedését (27), változatlanságát (27), illetve csökkenését (28) is. Humán szívelégtelen szívizom-mintákon végzett vizsgálatok megtartott Ca2+-áramot találtak a miokardiális LTCC kifejeződésének csökkenése ellenére, amit a csatorna fokozott foszforilációjával magyaráztak (28). Rágcsálókban a Ca2+-homeosztázisért felelős fehérjék megváltozott expresszióját azonosították a BKH-ban fennálló fokozott befelé irányuló Ca2+-áram fő okaként (4). Hipotézisük szerint a SERCA2 expressziójának csökkenése közvetett módon hozzájárul az LTCC fokozott aktivitásához BKH fennállása esetén. A SERCA2 csökkenése ugyanis csökkent Ca2+ indukálta Ca2+-felszabaduláshoz vezet a szarkoplazmás retikulumból, amely következtében a citoplazma lokális Ca2+-koncentráció alacsony marad és így az LTCC csatorna Ca2+-függő inaktivációja lassúbbá válik. Vizsgálatunkban az LTCC expressziója változatlan maradt a BKH kialakulásának ellenére (5. B ábra). Azonban a SERCA2 kifejeződése erős tendenciát mutatott a csökkenés irányába (5. C ábra), ami tehát indirekt módon a mi modellünkben is hozzájárulhatott a QT-intervallum megnyúlásához.
Ugyanakkor a szűkület eltávolítása a Debanded csoportban az LVmass, a HW/TL és a CD értékeket szignifikáns mértékben csökkentette, igazolva ezáltal a BKH regresszióját (1. táblázat, 1. A, B és 3. B ábrák). A hipertrófia visszafejlődéséhez a QT-intervallum rövidülése társult (2., 3. A ábrák), amelynek hátterében a SERCA2-fehérje expressziója által mutatott erős növekedő tendencia is állhat (5. C ábra).
A nyomásterhelés megszüntetése védett a szív funkcionális dekompenzációjával szemben és meggátolta a PQ-intervallum megnyúlását.
Vizsgálatunkban a QT-intervallummal ellentétben a PQ-intervallum megnyúlása nem követte a szívizom-hipertrófia növekedésének mértékét, ellenben korrelált a szívfunkció romlásával (4. B ábra). Eredményeinkkel összhangban, csökkent balkamra-funkcióval rendelkező, pangásos szívelégtelenségben szenvedő nyulakon végzett korábbi vizsgálatok az pitvar-kamrai junkció megnyúlásáról, bizonyos ioncsatornák expressziós szintű változásairól és következményesen a PR-intervallum megnyúlásáról számolnak be (12).
Aortaszűkített állatainkban a pumpafunkció romlását a 9. hét elteltével az EF és a FS szignifikáns csökkenése jelezte (2. táblázat, 3. B ábra). Emellett a LVEDP és LW/TL hányados értékeiben megfigyelt növekvő tendencia bizonyítékul szolgált a diasztolés diszfunkció és a visszaható szívelégtelenség fennállására. A szívfunkció romlását jelző paraméterek (csökkent EF, FS és növekedett LW/TL hányados) szoros összefüggést mutattak a PQ-távolság megnyúlásával, amely szintén a 9. héttől vált megfigyelhetővé. Ebből kifolyólag feltételezhetjük, hogy kisállat-modellünkben a csökkent bal kamrai teljesítmény miatt másodlagosan megnövekedett bal pitvari nyomásterhelés hozzájárulhatott a PQ-intervallum megnyúlásához.
Ugyanakkor a nyomásterhelés megszüntetése eredményesen előzte meg a BK funkcionális romlását és meggátolta a PQ-intervallum meghosszabbodását (3. C, D ábrák és 2. táblázat).
A nyomásterhelés megszüntetése megelőzte a QRS-komplexum kiszélesedését, csökkentette a miokardiális fibrózis mértékét és helyreállította a Cx43 expresszióját
A széles QRS-komplexum a súlyosan szívelégtelen betegek gyakran diagnosztizált EKG eltérése (9), amely a halálozás pontos előrejelzőjének bizonyult (29). A QRS-komplexum kiszélesedésének hátterében a kamrák elektromos vezetőképességének lassulása áll. Az elektromos impulzus szívizomsejtek közti továbbítását fiziológiás körülmények fennállása esetén a következő két tényező biztosítja.
Az optimális ingerületvezetéshez egyrészt szükség van az egyes szívizomsejteket összekötő interkalált lemezeken található kis ellenállású intercelluláris ioncsatornák megfelelő denzitására, amelyek felépítésében a Cx43 fehérje kitüntetett szerepet játszik (30). Ebből adódóan a Cx43 szerepe a szívelégtelenség talaján kialakuló aritmiákban intenzív kutatások tárgyát képezi. Expressziójának csökkenését számos különböző etiológiájú szívelégtelen beteg szívéből származó mintán kimutatták (31, 32). Ugyanakkor kompenzált BKH-ban megtartott vagy akár fokozott Cx43 fehérje-expressziót írtak le (31). Mindemellett aortaszűkített patkányokon végzett kísérletekben a Cx43 kóros, egyenlő sejtfelszíni eloszlását találták, ami az interkalált lemezeken csökkent koncentrációt eredményezett (33). A mi kísérletünkben a Cx43 szívizomszöveti expressziójának erősen csökkenő tendenciáját találtuk a 6. és 12. héten, bár ez nem érte el a szignifikancia küszöbét (5. A ábra).
Másrészt a szívizomsejtek közti kapcsolatot az intersticiális fibrózis mértéke is befolyásolja (3). Bár a kollagén struktúra adja a szív vázát, az extracelluláris mátrix túlzott felhalmozódása a sejtek közti kapcsolat zavarát idézheti elő, ami károsodott ingerületvezetést eredményezhet (10). Vizsgálatunkban a fokozott nyomásterhelés hatására csak a 12. hét elteltével találtunk jelentősen fokozott miokardiális fibrózist.
Állatainkban a Cx43 expressziója nem, viszont a kollagén felhalmozódás mértéke szignifikáns korrelációt mutatott a QRS-komplexum kiszélesedésével. Ezek alapján feltételezhetjük, hogy kísérletünkben a miokardiális fibrózis növekedése járult hozzá leginkább a megfigyelt QRS-kiszélesedéshez.
Másrészről a nyomásterhelés megszüntetése megelőzte a myocardium fibrotikus elváltozásait (1. A, C ábrák) és visszaállította a Cx43 expresszióját, ami együttesen védelmet biztosított a QRS-komplexum kiszélesedése ellen, és megtartotta a normális vezetést.
Következtetések
Vizsgálatunkban a fokozott nyomásterhelés kiváltotta BKH-t a QT- és PQ-intervallumok megnyúlása és a QRS-komplexum kiszélesedése jellemezték. Az EKG-eltérések a BKH kifejlődésének funkcionálisan különböző stádiumaiban jelentek meg, amelyek hátterében egyedi patomechanizmusokat azonosítottunk. A QT-intervallum megnyúlása jól korrelált a szívizom-hipertrófia kialakulásával, így már annak kompenzált stádiumában is jelen volt. A QT-intervallum megnyúlása mögött a SERCA2 szívizomszöveti kifejeződésének csökkenését azonosítottuk, mint fő molekuláris mechanizmust. A PQ-intervallum meghosszabbodása továbbá összefüggést mutatott a BKH funkcionális dekompenzációjával. Mindemellett kimutattuk, hogy a QRS kiszélesedéséért elsősorban a miokardiális fibrózis fokozódása, és nem a Cx43 expressziójának csökkenése tehető felelőssé.
A nyomásterhelés megszüntetése ugyanakkor maga után vonta a BKH szignifikáns regresszióját és a SERCA2 expressziójának helyreállását, amely folyamatok együtt jártak a QT-intervallum megrövidülésével. Továbbá a nyomásterhelés megszüntetése megőrizte a BK funkcionalitását és ez által védett a PQ-intervallum megnyúlásával szemben is. Végül a csökkent miokardiális fibrózis és a fokozott Cx43 expresszió kivédte a QRS kiszélesedését is.
Limitációk
Vizsgálatainkat kizárólag fiatal hím patkányokon végeztük. Így további kutatások szükségesek a kor és a nem befolyásoló hatásának, illetve a fajok közti esetleges különbségek tisztázására.
Bár az elektrofiziológiai fundamentumok közösek a patkányok és emberek közt, bizonyos különbségek, úgymint a magasabb szívfrekvencia, rövidebb AP-időtartam, és a repolarizációért felelős K+-csatornák eltérő eloszlása, struktúrája és funkciója, említést érdemelnek.
Annak ellenére, hogy az AB a nyomásterhelés okozta BKH elfogadott modellje, az abdomiális aorta műtéti beszűkítése következtében a vesék hipoperfúziója alakulhat ki, ami a renin–angiotenzin–aldoszteron-rendszer aktiválódásával, így volumenretencióval járhat (34). Tehát nem zárható ki, hogy a modellünkben kifejlődött BKH-hoz volumenterhelés is hozzájárult.
Bemutatott kísérletes eredményeink direkt átültetése a klinikai gyakorlatba a fentebb ismertetett különbségek miatt nem lehetséges. Ugyanakkor vizsgálatunk olyan fontos összefüggésekre világít rá a reverz elektromos remodelláció során, mely megfigyelések alapjául szolgálhatnak további humán vizsgálatoknak.
Köszönetnyilvánítás
Jelen munkánkat a Nemzeti Kutatási, Fejlesztési és Innovációs Hivatal – NKFIH (NVKP-16-1-2016-0017, „Nemzeti Szívprogram”); az Emberi Erőforrások Minisztériuma ÚNKP-17-3-I-6-SE kódszámú Új Nemzeti Kiválóság Programja (R.M.), a Magyar Tudományos Akadémia Bolyai János Kutatási Ösztöndíja (R.T.), a Baden-Württemberg tartomány és a Ruprecht-Karls Egyetem Orvostudományi kara (SK-I); támogatta. Elismerés illeti Patricia Kraft, Tobias Mayer, Karin Sonnenberg, Lutz Hoffmann és Gregor Viktória munkáját.
Irodalom
1. Ruppert M, Korkmaz-Icoz S, Li S, Merkely B, Karck M, Radovits T, et al. Reverse electrical remodeling following pressure unloading in a rat model of hypertension-induced left ventricular myocardial hypertrophy. Hypertens Res 2017; 40: 637–645. doi: https://doi.org/10.1038/hr.2017.1
2. McLenachan JM, Henderson E, Morris KI, Dargie HJ. Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med 1987; 317: 787–792. doi: https://doi.org/10.1056/NEJM198709243171302
3. Wolk R. Arrhythmogenic mechanisms in left ventricular hypertrophy. Europace 2000; 2: 216–223. doi: https://doi.org/10.1053/eupc.2000.0110
4. Ahmmed GU, Dong PH, Song G, Ball NA, Xu Y, Walsh RA, et al. Changes in ca(2+) cycling proteins underlie cardiac action potential prolongation in a pressure-overloaded guinea pig model with cardiac hypertrophy and failure. Circ Res 2000; 86: 558–570. doi: https://doi.org/10.1161/01.RES.86.5.558
5. Teunissen BE, Jongsma HJ, Bierhuizen MF. Regulation of myocardial connexins during hypertrophic remodeling. Eur Heart J 2004; 25: 1979–1989. doi: https://doi.org/10.1016/j.ehj.2004.08.007
6. Tomita F, Bassett AL, Myerburg RJ, Kimura S. Diminished transient outward currents in rat hypertrophied ventricular myocytes. Circ Res 1994; 75: 296–303. doi: https://doi.org/10.1161/01.RES.75.2.296
7. Guo W, Li H, London B, Nerbonne JM. Functional consequences of elimination of i(to,f) and i(to,s): Early after depolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking kv1.4 and expressing a dominant-negative kv4 alpha subunit. Circ Res 2000; 87: 73–79. doi: https://doi.org/10.1161/01.RES.87.1.73
8. Hill JA. Hypertrophic reprogramming of the left ventricle: Translation to the ecg. J Electrocardiol 2012; 45: 624–629. doi: https://doi.org/10.1016/j.jelectrocard.2012.08.003
9. Kashani A, Barold SS. Significance of QRS complex duration in patients with heart failure. J Am Coll Cardiol 2005; 46: 2183–2192. doi: https://doi.org/10.1016/j.jacc.2005.01.071
10. Tanaka-Esposito C, Varahan S, Jeyaraj D, Lu Y, Stambler BS. Eplerenone-mediated regression of electrical activation delays and myocardial fibrosis in heart failure. J Cardiovasc Electrophysiol 2014; 25: 537–544. doi: https://doi.org/10.1111/jce.12390
11. Wang Z, Kutschke W, Richardson KE, Karimi M, Hill JA. Electrical remodeling in pressure-overload cardiac hypertrophy: Role of calcineurin. Circulation 2001; 104: 1657–1663. doi: https://doi.org/10.1161/hc3901.095766
12. Nikolaidou T, Cai XJ, Stephenson RS, Yanni J, Lowe T, Atkinson AJ, et al. Congestive heart failure leads to prolongation of the pr interval and atrioventricular junction enlargement and ion channel remodelling in the rabbit. PLoS One 2015; 10: e0141452. doi: https://doi.org/10.1371/journal.pone.0141452
13. Gao XM, Kiriazis H, Moore XL, Feng XH, Sheppard K, Dart A, et al. Regression of pressure overload-induced left ventricular hypertrophy in mice. American journal of physiology. Heart and circulatory physiology 2005; 288: H2702–2707. doi: https://doi.org/10.1152/ajpheart.00836.2004
14. Ruppert M, Korkmaz-Icoz S, Li S, Nemeth BT, Hegedus P, Brlecic P, et al. Myocardial reverse remodeling after pressure unloading is associated with maintained cardiac mechano-energetics in a rat model of left ventricular hypertrophy. American journal of physiology. Heart and circulatory physiology 2016; 311: H592–603. doi: https://doi.org/10.1152/ajpheart.00085.2016
15. Rials SJ, Wu Y, Ford N, Pauletto FJ, Abramson SV, Rubin AM, et al. Effect of left ventricular hypertrophy and its regression on ventricular electrophysiology and vulnerability to inducible arrhythmia in the feline heart. Circulation 1995; 91: 426–430. doi: https://doi.org/10.1161/01.CIR.91.2.426
16. Olah A, Nemeth BT, Matyas C, Hidi L, Lux A, Ruppert M, et al. Physiological and pathological left ventricular hypertrophy of comparable degree is associated with characteristic differences of in vivo hemodynamics. American journal of physiology. Heart and circulatory physiology 2016; 310: H587–597. doi: https://doi.org/10.1152/ajpheart.00588.2015
17. Nemeth BT, Matyas C, Olah A, Lux A, Hidi L, Ruppert M, et al. Cinaciguat prevents the development of pathologic hypertrophy in a rat model of left ventricular pressure overload. Sci Rep 2016; 6: 37166. doi: https://doi.org/10.1038/srep37166
18. Kim HL, Kim YJ, Kim KH, Lee SP, Kim HK, Sohn DW, et al. Therapeutic effects of udenafil on pressure-overload cardiac hypertrophy. Hypertens Res 2015; 38: 597–604. doi: https://doi.org/10.1038/hr.2015.46
19. Korkmaz-Icoz S, Atmanli A, Radovits T, Li S, Hegedus P, Ruppert M, et al. Administration of zinc complex of acetylsalicylic acid after the onset of myocardial injury protects the heart by upregulation of antioxidant enzymes. J Physiol Sci 2016; 66: 113–125. doi: https://doi.org/10.1007/s12576-015-0403-6
20. Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, et al. Echocardiographic assessment of left ventricular hypertrophy: Comparison to necropsy findings. Am J Cardiol 1986; 57: 450–458. doi: https://doi.org/10.1016/0002-9149(86)90771-X
21. Pombo JF, Troy BL, Russell RO, Jr. Left ventricular volumes and ejection fraction by echocardiography. Circulation 1971; 43: 480–490. doi: https://doi.org/10.1161/01.CIR.43.4.480
22. Kmecova J, Klimas J. Heart rate correction of the QT duration in rats. Eur J Pharmacol 2010; 641: 187–192. doi: https://doi.org/10.1016/j.ejphar.2010.05.038
23. Korkmaz-Icoz S, Vater A, Li S, Lehner A, Radovits T, Hegedus P, et al. Mild type 2 diabetes mellitus improves remote endothelial dysfunction after acute myocardial infarction. J Diabetes Complications 2015; 29: 1253–1260. doi: https://doi.org/10.1016/j.jdiacomp.2015.06.012
24. Boulaksil M, Noorman M, Engelen MA, van Veen TA, Vos MA, de Bakker JM, et al. Longitudinal arrhythmogenic remodeling in a mouse model of longstanding pressure overload. Neth Heart J 2010; 18: 509–515.
25. Klimas J, Kruzliak P, Rabkin SW. Modulation of the qt interval duration in hypertension with antihypertensive treatment. Hypertens Res 2015; 38: 447–454. doi: https://doi.org/10.1038/hr.2015.30
26. Farraj AK, Hazari MS, Cascio WE. The utility of the small rodent electrocardiogram in toxicology. Toxicol Sci 2011; 121: 11–30. doi: https://doi.org/10.1093/toxsci/kfr021
27. Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, et al. Increased availability and open probability of single l-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998; 98: 969–976. doi: https://doi.org/10.1161/01.CIR.98.10.969
28. Chen X, Piacentino V, 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res 2002; 91: 517–524. doi: https://doi.org/10.1161/01.RES.0000033988.13062.7C
29. Iuliano S, Fisher SG, Karasik PE, Fletcher RD, Singh SN, Department of Veterans Affairs Survival Trial of Antiarrhythmic Therapy in Congestive Heart F. Qrs duration and mortality in patients with congestive heart failure. Am Heart J 2002; 143: 1085–1091. doi: https://doi.org/10.1067/mhj.2002.122516
30. Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res 2004; 62: 228–232. doi: https://doi.org/10.1016/j.cardiores.2003.11.013
31. Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res 2004; 62: 426–436. doi: https://doi.org/10.1016/j.cardiores.2003.12.010
32. Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klovekorn WP, et al. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem 2003; 242: 135–144. doi: https://doi.org/10.1023/A:1021154115673
33. Emdad L, Uzzaman M, Takagishi Y, Honjo H, Uchida T, Severs NJ, et al. Gap junction remodeling in hypertrophied left ventricles of aortic-banded rats: Prevention by angiotensin ii type 1 receptor blockade. J Mol Cell Cardiol 2001; 33: 219–231. doi: https://doi.org/10.1006/jmcc.2000.129
34. Massart PE, Donckier J, Kyselovic J, Godfraind T, Heyndrickx GR, Wibo M. Carvedilol and lacidipine prevent cardiac hypertrophy and endothelin-1 gene overexpression after aortic banding. Hypertension 1999; 34: 1197–1201. doi: https://doi.org/10.1161/01.HYP.34.6.1197