Qualitative and quantitative assessement of ECG characteristics in patients with Andersen–Tawil syndrome
█ Original article
DOI: 10.26430/CHUNGARICA.2019.49.5.344
Authors:
Borbás János1, Takács Hedvig1, Környei László2, Katona Márta3, Ördög Balázs4
1Szegedi Tudományegyetem, II. sz. Belgyógyászati Klinika és Kardiológiai Központ, Szeged
2Gottsegen György Országos Kardiológiai Intézet, Budapest
3Szegedi Tudományegyetem, Gyermekgyógyászati Klinika, Szeged
4Szegedi Tudományegyetem, Farmakológiai és Farmakoterápiai Intézet, Szeged
Summary
Background: Andersen–Tawil syndrome (ATS) is a multisystem disorder mainly caused by mutations in KCNJ2 gene which encodes the main pore-forming protein of the inward rectifier K+ channel, Kir2.1. The disease is characterized by ventricular arrhythmias, periodic paralysis, dysmorphic features and specific ECG alterations.
Objective: Our aim was to qualitatively and quantitatively analyse typical ECG characteristics, described in the literature, in our patient cohort with ATS in comparison with that of observed in patients with the long QT syndrome (LQTS).
Patients and methods: Seven patients (6 females, 1 male, avg. age 25.4±11 yrs) with genetically confirmed ATS (three patients with p.Arg218His, one patient each with p.Arg312Glu, p.del302Val, p.Glu293Lys and p.Met307Ile KCNJ2 mutations) were examined. Quantitative ECG parameters [premature ventricular beats (PVB)/bigeminy, couplet, NSVT] and corrected QT and QU intervals, the duration and amplitude of the U waves were manually measured on good quality 12-lead resting ECG recordings. Data were compared to that of age- and sex-matched control patients with long QT syndrome (LQTS).
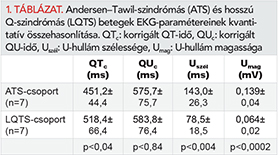
Results: Frequent ventricular extra-systoles (or bigeminy) were present in 71% (5/7) of ATS patients. Couplets or NSVT was observed in 57% (4/7) of ATS patients. As for quantitative ECG parameters corrected QT interval was significantly shorter in ATS patients compared to LQTS patients (451.2±44.4 vs. 518.4±66.4 ms, p<0.04), while corrected QU interval did not differ significantly (575.7±75.7 vs. 583.8±76.4 ms, p<0.84). The QTc was prolonged only in 43% (3/7) of ATS patients. The U waves were significantly longer (U wave duration: 143.0±26.3 vs. 78.5±18.5 ms, p<0.004) and taller (U vawe amplitude: 0.139±0.04 vs. 0.064±0.02 mV, p<0.0002) in ATS patients.
Conclusion: Patients with ATS usually exhibit typical ECG changes representing frequent ventricular ES, bigeminy or NSVT. Prolongation of the corrected QT interval is not typical and QTc may be in the normal range. U waves are more prominent, being longer and taller in these patients.
ISSUE: CARDIOLOGIA HUNGARICA | 2019 | VOLUME 49, ISSUE 5
Összefoglalás
Háttér: Az Andersen–Tawil-szindróma (ATS) egy több szervrendszert érintő kórkép, amelyet az esetek jelentős részében a befelé egyenirányító K+-csatorna fő pórusformáló fehérjéjének, a Kir2.1-fehérje kódolásáért felelős KCNJ2-gén mutációi okoznak. A betegséget kamrai aritmiák, periodikus paralízis, fejlődési rendellenességek és specifikus EKG-eltérések jellemezik.
Célkitűzés: Célunk az volt, hogy ATS-betegek irodalomból ismert EKG-eltéréseinek kvalitatív és kvantitatív paramétereit elemezzük saját ATS-betegcsoportunkban, hosszú QT-szindrómás (LQTS) betegek EKG-paramétereivel való összehasonlításban.
Betegek és módszerek: Hét (6 nő, 1 férfi., átlagéletkor 25,4±11 év) genetikai vizsgálattal igazolt ATS (három p.Arg218His-mutáció, további egy-egy p.Arg312Glu, p.del302Val, p.Glu293Lys és p.Met307Ile KCNJ2-mutáció) beteget vizsgáltunk. A 12 elvezetéses nyugalmi EKG-felvételeket kvalitatív [gyakori kamrai extraszisztolék (ES)/bigeminia, couplet, NSVT jelenléte], illetve kvantitatív szempontból értékeltük (korrigált QT- és QU-intervallum, az U-hullám időtartama és amplitúdója). Mérési eredményeinket kor- és nem szerint illesztett kontroll, LQTS-betegek adataival hasonlítottuk össze.
Eredmények: Gyakori kamrai extraszisztolé (vagy bigeminia) az ATS esetek 71%-ában (5/7) volt jelen. Couplet vagy NSVT az ATS esetek 57%-ában (4/7) volt megfigyelhető. Kvantitatív EKG-paraméterek tekintetében a korrigált QT-intervallum szignifikánsan rövidebb volt az ATS-betegekben az LQTS-betegekhez képest (451,2±44,4 vs. 518,4±66,4 ms, p<0,04), míg a korrigált QU-intervallum nem különbözött szignifikánsan (575,7±75,7 vs. 583,8±76,4 ms, p<0,84). A QTc- az ATS-esetek mindössze 43%-ában (3/7) volt megnyúlt tartományban. Az U-hullámok szignifikánsan hosszabbnak (U-hullám időtartam: 143,0±26,3 vs. 78,5±18,5 ms, p<0,004) és magasabbnak (U-hullám amplitúdó: 0,139±0,04 vs. 0,064±0,02 mV, p<0,0002) bizonyultak az ATS-betegek esetében.
Következtetés: Andersen–Tawil-szindrómás betegekben általában jellegzetes EKG-elváltozásokat láthatunk, mint pl. a gyakori kamrai ES, bigeminia vagy NSVT. A korrigált QT-intervallum kóros tartományba való megnyúlása nem minden esetben jellemző. Az U-hullám prominensebb, hosszabb és magasabb ezekben a betegekben.
Bevezetés
Az Andersen–Tawil-szindróma (ATS) a familiáris szívizombetegségekhez [hipertrófiás cardiomyopathia (1, 2, 3), Danon-betegség (4), Fabry-betegség (5) stb.] és a familiáris ioncsatorna-betegségekhez hasonlóan [hosszú QT-szindróma (6), Timothy-szindróma (7), familiáris bradycardia (8) stb.] monogénes, mendeli öröklődést mutató ritka kórkép. A betegséget az alapvetően az izomrostokban azonosítható, befelé irányító, egyenirányú (inward rectifyer) káliumion-csatorna fő pórusformáló fehérjéjét, a Kir2.1-fehérjét kódoló gént, a KCNJ2-gént érintő mutációk okozzák (9).
Az ATS ritka kórkép, amelyet periodikus paralízis, kamrai aritmiák és fejlődési rendellenességek triásza jellemez. A fejlődési rendellenességek scoliosis, clinodactylia és syndactylia, a testméret és súly elmaradása, valamint craniofacialis anomáliák, mint a hipertelorizmus, széles orrnyereg, mandibuláris hipoplázia, bilaterális ptosis, vékony haj, fogfejlődési malformációk, mandibuláris és maxilláris hipoplázia formájában nyilvánulnak meg (10, 11, 12, 13). Az ATS-ben kialakuló periodikus paralízist hirtelen kezdődő, néhány perctől akár néhány napig változó intervallumra is kiterjedő izomgyengeség vagy paralízis jellemzi, amely normo-, hypo- és hyperkalaemia mellett egyaránt kialakulhat.
Az ATS kardiális manifesztációi közül az EKG-n megfigyelhető eltérések a leggyakoribbak és leginkább figyelmet keltőek. Egyes betegekben enyhe QTc-megnyúlás észlelhető, de a QUc-tartam jelentős megnyúlása és a kifejezett U-hullámok jelenléte a legjellemzőbb. Az U-hullámok szélesek és magasak, leginkább a precordiális V2–3-elvezetésekben (14, 15). Az ATS-ben megfigyelhető aritmiák között a gyakori kamrai extraszisztolék, coupletek és típusos formában bidirekcionális polimorf kamrai tachycardiák (VT) a legjellemzőbbek, amelyek akár tartósak, akár nem tartósak lehetnek (14).
A betegség Magyarországon is dokumentált, az első, genetikai vizsgálattal is igazolt ATS esetleírást munkacsoportunk közölte (16). Jelen vizsgálatunkban célunk az volt, hogy ATS-betegek EKG-regisztrátumainak kvalitatív és kvantitatív paramétereit elemezzük, amelyeket hosszú QT-szindrómás (LQTS) betegek EKG-paramétereivel hasonlítottunk össze.
Betegek és módszerek
Betegek
Munkánk során öt családhoz tartozó hét (6 nő, 1 férfi., átlagéletkor a diagnóziskor 25,4±11 év), konszekutív Andersen–Tawil-szindrómás beteget vizsgáltunk. A betegség klinikai diagnózisát a szakirodalomban javasoltak szerint állítottuk fel, az alábbi három klinikai kritérium közül legalább kettő megléte alapján:
- periódikus paralízis előfordulása;
- tüneteket okozó aritmiák vagy megnagyobbodott U-hullámok, kamrai ektópiák, megnyúlt QTc- vagy QUc-intervallum igazolása;
- jellemző arceltérések, fogászati eltérések, kis kéz és láb méret mellett legalább két további jellemző észlelése: alacsonyan álló fülek, szélesen álló szemek, kis mandibula, kisujjat érintő clinodactylia vagy 2-3 lábujjat érintő syndactylia (17).
Minden beteg esetében genetikai vizsgálat is igazolta a betegséget, amely szerint mind a hét esetben patogén mutációt volt igazolható a KCNJ2-génben (három p.Arg218His-mutáció, további egy-egy p.Arg312Glu, p.del302Val, p.Glu293Lys és p.Met307Ile KCNJ2-mutáció).
Az ATS-betegek EKG-jellemzőit hosszú QT-szindrómában szenvedő betegek EKG-jellemzőivel hasonlítottuk össze. Az összehasonlításra használt LQTS-betegek EKG-it Intézetünk adatbázisából választottuk ki, random módon, a betegcsoport esetszámával 1:1 arányban. Az illesztés alapjául az szolgált, hogy kor- és nemi eloszlás szerint a két betegcsoport azonos legyen. Ennek megfelelően az LQTS-betegcsoportban 6 nő és 1 férfi szerepelt, diagnóziskori átlagéletkoruk 26,0±9 év volt.
A két betegcsoport gyógyszeres terápiájában lényeges különbség nem volt, az ATS-betegcsoportban 7/7, az LQTS-betegcsoportban 6/7 beteg állt béta-blokkoló terápia alatt. Az ATS-betegcsoportban 2/7 betegben, az LQTS-betegcsoportokban 3/7 betegben történt ICD-implantáció.
EKG-analízis
Az ATS- és LQTS-betegeknél klinikai kivizsgálásuk során standard nyugalmi 12 elvezetéses EKG-felvételek készültek. Az EKG-regisztrátumokat kvalitatív és kvantitatív szempontból vizsgáltuk. A kvalitatív eltérések között a kamrai extraszisztolék (ES)/bigeminia jelenlétét, illetve a coupletek és NSVT jelenlétét értékeltük. Az NSVT-t három vagy több ütésből álló, >100/min frekvenciájú és <30 másodpercig tartó ritmuszavarként definiáltuk. A bidirekcionális VT-t a QRS-tengelyének 180°-kal való, ütésről ütésre való váltakozásaként határoztuk meg. A kvantitatív EKG-paraméterek között a korrigált QT (QTc) és QU (QUc) intervallumot, az U-hullám időtartamát és az U-hullám amplitúdóját mértük, a V2- vagy V3-elvezetésben. Utóbbiak manuális mérés alapján lettek meghatározva. A szívfrekvenciára való korrekció a Bazett-formula alapján történt.
Eredmények
A korra és nemre való illesztésnek megfelelően a két betegcsoport kor (25,4±11,6 vs. 26,0±9,3 év; p=0,9249) és nem eloszlását illetően (6 nő/1 férfi vs. 6 nő/1 férfi; p=1,000) szignifikáns különbség nem volt. Hasonlóképp nem volt szignifikáns különbség a betegek nyugalmi szívfrekvenciáit illetően (68,4±5,6 vs. 71,2±3,4/min).
ATS-betegek EKG-regisztrátumainak kvalitatív értékelése
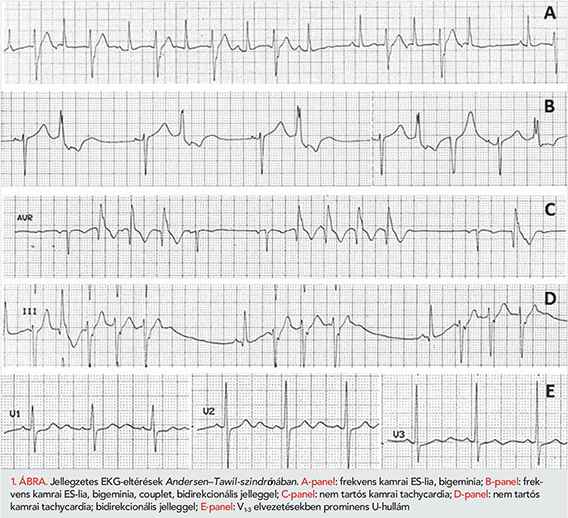
Gyakori kamrai extraszisztolé (vagy bigeminia) az Andersen–Tawil-szindrómás esetek 71%-ában (5/7) volt jelen (1. ábra, A- és B-panel) Couplet vagy NSVT- az ATS-betegcsoportban 57%-ban (4/7) volt megfigyelhető (1. ábra, B–D-panel). Ez egy esetben volt típusos bidirekcionális NSVT (1. ábra D-panel), további három esetben 180 o-os változó polaritást mutató couplet formájában jelentkezett (1. ábra B-panel).
ATS- és LQTS-betegek EKG-paramétereinek kvantitatív elemzése
Az ATS- és LQTS-betegek QTc, QUc, U-hullám szélesség és magasság paramétereit az 1. táblázatban tüntettük fel. Kvantitatív EKG-paraméterek tekintetében a korrigált QT-intervallum szignifikánsan rövidebb volt az ATS-betegekben az LQTS-betegekhez képest (451,2±44,4 vs. 518,4±66,4 ms, p<0,04), míg a korrigált QU-intervallum nem különbözött szignifikánsan (575,7±75,7 vs. 583,8±76,4 ms, p<0,84). A QTc- az ATS-esetek mindössze 43%-ban (3/7) volt megnyúlt tartományban (nők esetében >460 ms, férfiak esetében >450 ms). Az U-hullámok szignifikánsan hosszabbnak (U-hullám időtartam: 143,0±26,3 vs. 78,5±18,5 ms, p<0,004) és magasabbnak (U-hullám amplitúdó: 0,139±0,04 vs. 0,064±0,02 mV, p<0,0002) bizonyultak az ATS-betegek esetében (1. ábra, E-panel).
Megbeszélés
Munkánk során magyarországi Andersen–Tawil-szindrómás betegek EKG-eltéréseinek kvalitatív és kvantitatív elemzését végeztük el. Eredményeink szerint, az irodalomban közölt korábbi megfigyelésekhez hasonlóan, ATS-betegekben jellegzetes EKG-elváltozásokat láthatunk, mint pl. a gyakori kamrai extraszisztolé, bigeminia vagy NSVT, az esetek egy részében bidirekcionális formában. A korrigált QT-intervallum gyakran rövidebb, mint az a hosszú QT-szindrómás betegekben megfigyelhető, és akár a normál tartományban is lehet. Az U-hullám prominensebb, hosszabb és magasabb ezekben a betegekben.
Jól ismert és klinikai fontossággal bír az a tény, hogy testfelszíni EKG egyes eltérései, illetve a T-hullám formája eltérő, és adott esetben jellemző lehet a különböző LQT-alcsoportokban. Az LQT1-alcsoportban viszonylag hamar megjelenő, széles alapú, megnyúlt T-hullám a jellemző; míg az LQT2-alcsoportot a T-hullámok jelentős amplitúdó csökkenése jellemzi, bifid, hasadt, konfluáló U-hullámra hasonlító, több hullámkomponensből álló T-hullám megjelenésével, elsősorban a mellkasi elvezetésekben. Az LQT3-alcsoport jellemző EKG-elváltozása az ST-szakasz megnyúlása, azaz a QT-megnyúlást a magában normális időtartamú és amplitúdójú T-hullám késői megjelenése okozza (18).
Az ATS-ben megfigyelhető EKG jellemzőket Zhang és munkatársai vizsgálták elsőként (15). A vizsgált populációban (96 ATS-es beteg) a QTc-megnyúlás mértéke nem volt kifejezett (20 ms), a QTc medián értéke (440 ms) a normál tartományon belül volt. A betegek csupán 17%-ának volt QTc-értéke 460 ms-ot meghaladó. A betegek 91%-ában a T-U-hullám morfológiája, különösképp a megnagyobbodott U-hullám karakterisztikus volt. A betegek egy részénél a T-hullám leszálló szára megnyúlt, az U-hullám bifázisos volt. Eredményeink fenti megfigyelésekkel jól korrelálnak.
Az Andersen–Tawil-szindrómás betegek EKG jellegzetességeinek standardizálására tettek kísérletet Kukla és munkatársai, akik az Andersen–Tawil-szindrómás betegek EKG-analízise során írtak le diagnosztikus jellemzőket (19):
- tachycardiás ATS-betegekben a P-hullám és U-hullám „összecsúszásakor” p-pulmonale morfológia alakul ki és a P-hullám magassága meghaladhatja a 3 mm-t;
- az U-hullám jelenlétét figyelembe véve és a számításokból azt kihagyva a betegek QTc-tartama a normálintervallumba esik;
- VES-t követő első szívciklusnál a T- és U-hullám fuzionálhat, így pszeudo-LQTS-morfológiát mutathat;
- V2–3-elvezetésekben előfordulhat U-hullám, amelynek amplitúdóját (egészséges kontrollcsoporttal ellentétben) a tachycardia tovább növelheti;
- adrenalin-provokációs teszt hatására az U/T-hullám aránya <1-ről >1-re változik.
Az ATS-ben megfigyelhető ritmuszavarok között a gyakori kamrai extraszisztolék, esetleges bigeminia mellett a bidirekcionális jellegű polimorf kamrai tachycardia (VT) a legjellemzőbb.
Típusos esetben a VT nem-tartós jellegű, és relatíve lassú, ≤150/min frekvenciával. Bár a VT nem gyors és általában jól tolerált, az ES- és VT-epizódok mennyisége tachycardia-indukált cardiomyopathiához vezethet. Fontos az ATS-ben, digitálisz toxicitás esetén és (20) és katekolaminerg polimorf kamrai tachycardiában (CPVT) (21) észlelhető bidirekcionális VT elkülönítése egymástól, hiszen az ATS és CPVT prognózisa és terápiája eltér egymástól. Bár az ES- és VT-epizódok mennyisége ATS-ben jellemzően nagy, de a CPVT-betegekkel ellentétben életveszélyes kamrai ritmuszavarba való átmenet nem jellemző (15). A fizikai terhelés – CPVT-betegekkel ellentétben – nem indukál ritmuszavart ATS-betegekben, sőt egyes közlemények szerint a terhelés el is nyomhatja a ritmuszavart. A két betegség genetikai vizsgálattal különíthető el, a CPVT-esetek legtöbbje és az ATS-esetek mintegy 60%-a diagnosztizálható genetikai módszerekkel.
Következtetések
Andersen–Tawil-szindrómás betegekben általában jellegzetes EKG-elváltozásokat láthatunk, mint pl. a gyakori kamrai ES, bigeminia vagy NSVT. A korrigált QT-intervallum megnyúlása nem jellemző, és a QTc akár a normál tartományban is lehet. Az U-hullám prominens, hosszabb és magasabb ezekben a betegekben.
Köszönetnyilvánítás
A munka a „Ritka betegségek patogenezisének kutatása, új diagnosztikai és terápiás eljárásokat megalapozó fejlesztések” (GINOP-2.3.2-15-2016-00039) és a „Életet veSzélyezTető Akut megbetegedések súlYossági és hALálozási mutatólnak jaVítása transzlációs orvostudományi mEgközelítésben – STAY ALIVE” (GINOP-2.3.2-15-2016-00048) támogatásával készült.
Irodalom
1. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733–2779. doi: https://doi.org/10.1093/eurheartj/ehu284
2. Toth T, Nagy V, Faludi R, et al. The Gln1233ter mutation of the myosin binding protein C gene: Causative mutation or innocent polymorphism in patients with hypertrophic cardiomyopathy? Int J Cardiol 2011; 153(2): 216–9. doi: https://doi.org/10.1016/j.ijcard.2011.09.062
3. Orosz A, Baczko I, Nagy V, et al. Short-term beat-to-beat variability of the QT interval is increased and correlates with parameters of left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Can J Physiol Pharmacol 2015; 93(9): 765–72. doi: https://doi.org/10.1139/cjpp-2014-0526
4. Csanyi B, Popoiu A, Hategan L, et al. Identification of two novel LAMP2 gene mutations in Danon disease. Can J Cardiol 2016; 32(11): 1355.e23–1355.e30. doi: https://doi.org/10.1016/j.cjca.2016.02.071
5. Csányi B, Hategan L, Nagy V, et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease with a Predominant Cardiac Phenotype Int Heart J. 2017; 58(3): 454–458. doi: https://doi.org/10.1536/ihj.16–361
6. Csanády M, Kiss Z. Az elektrokardiogram QT-távolságának örökletes megnyúltsága, veleszületett süketség nélkül (Romano-Ward-syndroma). Orv Hetil 1972; 47: 2840–2843.
7. Sepp R, Hategan L, Bácsi A, et al. Timothy Syndrome 1 Genotype without Syndactyly and Major Extracardiac Manifestations. Am J Med Genet A 2017; 173(3): 784–789. doi: https://doi.org/10.1002/ajmg.a.38084.
8. Hategan L, Csányi B, Ördög B, et al. A novel ‘splice site’ HCN4 gene mutation, c.1737+1 G>T, causes familial bradycardia, reduced heart rate response, impaired chronotropic competence and increased short-term heart rate variability. Int J Cardiol 2017; 241: 364–372. doi: https://doi.org/10.1016/j.ijcard.2017.04.058
9. Raab-GrahamKF, Radeke CM, Vandenberg CA Molecular cloning and expression of a human heart inward rectifier potassium channel. Neuroreport 1994; 5: 2501–5. doi: https://doi.org/10.1097/00001756-199412000-00024
10. Klein R, Ganelin R, Marks JF, et al. Periodic paralysis with cardiac arrhythmia. J Pediatr 1963; 62: 371–85. doi: https://doi.org/10.1016/S0022-3476(63)80134-1
11. Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand 1971; 60: 559–64. doi: https://doi.org/10.1111/j.1651–2227.1971.tb06990.x
12. Donaldson MR, Jensen JL, Tristani-Firouzi M, et al. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 2003; 60: 1811–6. doi: https://doi.org/10.1212/01.WNL.0000072261.14060.47
13. Rajakulendran S, Tan SV, Hanna MG Muscle weakness, palpitations and a small chin: the Andersen–Tawil syndrome. Pract Neurol 2010; 10(4): 227–31. doi: https://doi.org/10.1136/jnnp.2010.217794
14. Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 2002; 110: 381–8. doi: https://doi.org/10.1172/jci15183
15. Zhang L, Benson DW, Tristani-Firouzi M, et al. Electrocardiographic features in Andersen–Tawil syndrome patients with KCNJ2 mutations: characteristic T–U-wave patterns predict the KCNJ2 genotype. Circulation 2005; 111: 2720–2726. doi: https://doi.org/10.1161/CIRCULATIONAHA.104.472498
16. Ördög B, Hategan L, Kovács M, et al. Identification and functional characterisation of a novel KCNJ2 mutation, Val302del, causing Andersen-Tawil syndrome. Can J Physiol Pharmacol 2015; 93(7): 569–75. doi: https://doi.org/10.1139/cjpp-2014-0527.
17. Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [Updated 2018 Jun 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019.
18. Zhang L, Timothy K, Vincent G, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome. ECG findings identify genotypes. Circulation 2000; 102: 2849–55.
19. Kukla P, Biernacka EK, Baranchuk A, et al. Electrocardiogram in Andersen-Tawil Syndrome. New Electrocardiographic Criteria for Diagnosis of Type-1 Andersen-Tawil Syndrome. Curr Cardiol Rev 2014; 10(3): 222–8. doi: https://doi.org/10.2174/1573403X10666140514102528
20. Piccini J, Zaas A Cases from the Osler medical service at Johns Hopkins University. Digitalis toxicity with bidirectional ventricular tachycardia. Am J Med 2003; 115: 70–71. doi: https://doi.org/10.1016/s0002-9343(03)00331-0
21. Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103: 196–200 doi: https://doi.org/10.1161/01.cir.103.2.196