Identification of a titin and desmoplakin double gene mutation in peripartum cardiomyopathy: genetic analysis of the first patient with heart transplantation performed in Szeged
█ Case report
DOI: 10.26430/CHUNGARICA.2020.50.2.132
Authors:
Csányi Beáta1, Bogáts Gábor1, Rudas László2, Babik Barna2, Nagy Viktória1, Tringer Annamária1, Hategan Lidia1, Borbás János1, Hegedűs Zoltán3,4,
Nagy István5,6, Sepp Róbert1
1Szegedi Tudományegyetem, II. sz. Belgyógyászati Klinika és Kardiológiai Központ, Szeged
2Szegedi Tudományegyetem, Aneszteziológiai és Intenzív Terápiás Intézet, Szeged
3Biofizikai Intézet, Szegedi Biológiai Központ, Szeged
4Pécsi Tudományegyetem, Biokémiai és Orvosi Kémiai Intézet, Pécs
5Biokémiai Intézet, Szegedi Biológiai Központ, Szeged
6Seqomics Biotechnológiai Kft.
Summary
Peripartum cardiomyopathy (PPC) is an idiopathic cardiomyopathy presenting with heart failure secondary to left ventricular systolic dysfunction towards the end of pregnancy or in the months following delivery, when no other cause of heart failure is found. Data support the observation that PPC and dilated cardiomyopathy (DCM) share a similar genetic background in a number of cases.
We performed genetic analysis of a 36-years-old female patient who underwent heart transplantation due to PPC in Szeged. The patient, with her second pregnancy, was admitted to our Institution at 38 weeks of gestation because of dyspnea. Echocardiography showed a dilated left ventricle with reduced left ventricular ejection fraction (EF: 30%). Symptoms of heart failure progressed despite of intensive treatment including levosimendan, dopamine, dobutamine and IABP support. The left ventricular function further deteriorated (EF: 14%), low cardiac output syndrome (CI: 1.4 l/min/m2, SV: 21 ml) developed, and mechanical ventilation was necessary because of hypoxia. Due to the life-threatening clinical situation, the only solution was urgent heart transplantation, which was carried out in Szeged, as the patient was unsuitable for transport. Family screening revealed that her mother had dilated cardiomyopathy. Based on this, familial cardiomyopathy was suspected, and genetic screening was performed.
Genotyping was performed by next generation sequencing, with a platform including 103 causative cardiomyopathy genes. Genetic screening detected 5 variants, and two of them were pathogenic with a truncation effect, one in the titin gene (TTN p.Arg13527Stop) and another in the desmoplakin gene (DSP p.Arg2284Stop). The pathogenic variants predispose to lose normal protein function either by protein truncation or nonsense-mediated mRNA decay. The p.Arg13527Stop variant in the TTN gene has been reported in association with DCM and the p.Arg2284Stop variant in the DSP gene has been published previously in association with ARVD/C and Carvajal-syndrome. So far, no case of these two pathogenic variants occurring together has been published.
Our case highlights the fact that some of the PPCs may have genetic origin. A family screening should be performed in each patient and genetic screening when suspicion arises.
ISSUE: CARDIOLOGIA HUNGARICA | 2020 | VOLUME 50, ISSUE 2
Összefoglalás
A peripartum cardiomyopathia (PPC) a terhesség végső időszakában vagy a szülést követő hónapokban kialakuló, balkamra-diszfunkció miatti szívelégtelenség képében megjelenő idiopathias cardiomyopathia, amelynek hátterében a szívelégtelenség más okai kizárhatók. Adatok utalnak arra, hogy a PPC-t az esetek egy részében genetikai eltérések okozzák, és ez a genetikai háttér átfedést mutat a dilatatív cardiomyopathiában (DCM) megfigyeltekkel.
Munkánkban PPC miatt Szegeden szívtranszplantáción átesett 36 éves nőbeteg genetikai analízisét végeztük el. A második terhességét viselő beteg terhessége 38. hetében került észlelésre nehézlégzés miatt, amelynek hátterében az echokardiográfiás vizsgálat tágult bal kamrát, csökkent balkamra-funkciót (EF: 30%) írt le. A szívelégtelenség tünetei levosimendan, dopamin, dobutamin adása, IABP keringéstámogatás ellenére progrediáltak, a balkamra-funkció tovább romlott (EF: 14%), alacsony perctérfogat (CI: 1,4 l/min/m2, SV: 21 ml) szindróma alakult ki, hypoxia miatt gépi lélegeztetés vált szükségessé. A válságos állapot miatt egyedüli megoldásként urgens szívtranszplantáció jött szóba, amelyet a beteg szállíthatatlan állapota miatt Szegeden végeztek el. Családszűrése során derült fény arra, hogy édesanyja dilatatív cardiomyopathiában szenved, amely alapján familiáris cardiomyopathia merült fel, és genetikai vizsgálat történt.
A genetikai vizsgálat újgenerációs szekvenálással történt, 103 cardiomyopathia-gént tartalmazó panel célzott újraszekvenálásával. A genetikai vizsgálat során összesen 5 variánst detektáltunk, amelyek közül a titingén (TTN) p.Arg13527Stop és a dezmoplakingén (DSP) p.Arg2284Stop variánsa bizonyult kóroki mutációnak. Mindkét variáns ismert a szakirodalomban, a TTN p.Arg13527Stop variáns DCM-ben szerepelt kóroki variánsként, míg a DSP p.Arg2284Stop variánst ARVC-vel és Carvajal-szindrómával hozták kapcsolatba. Eddig még nem publikáltak olyan estet, ahol ez a két patogén variáns együtt jelent volna meg.
Esetünk felhívja a figyelmet arra, hogy a peripartum cardiomyopathia az esetek egy részében genetikai eredetű lehet, ezért a PPC-betegek családszűrése és adott esetben genetikai vizsgálata lehet indokolt.
Bevezetés
A peripartum cardiomyopathia (PPC) az ESC Heart Failure Association legutóbbi definíciója szerint „a terhesség végső időszakában vagy a szülést követő hónapokban kialakuló, balkamra-diszfunkció miatti szívelégtelenség képében megjelenő idiopathiás cardiomyopathia, amelynek hátterében a szívelégtelenség más okai kizárhatók. A diagnózis kizáráson alapul. A bal kamra nem mindig tágult, de a bal kamrai ejekciós frakció majdnem minden esetben 45% alá csökken” (1). Bár a betegség az egész világon ismert, a világ egyes területein, pl. Nigériában és Haitin különösen magas az incidenciája. A betegség kialakulása szempontjából további rizikófaktort jelent a pre-eclampsia előfordulása és az előrehaladott anyai életkor. Bár a PPC patofiziológiája továbbra sem tisztázott részleteiben, a legutóbbi évtizedek kutatása a PPC-re hajlamos egyénekben vaszkulohumorális folyamatok jelentőségét hangsúlyozzák. Az érintett nők több mint felében a balkamra-diszfunkció normalizálódik, néhányukban krónikus cardiomyopathia alakul ki, és az eseteként kialakuló refrakter szívelégtelenség miatt mechanikus keringéstámogatás és/vagy szívtranszplantáció válik szükségessé. További potenciális komplikációként tromboembólia vagy ritmuszavar fordulhat elő. A PPC kezelése megegyezik a csökkent ejekciós frakciójú szívelégtelenség kezelésével, adott esetben a magzat jelentette kontraindikációk figyelembevételével. A PPC-ben specifikusan alkalmazható bromocriptinterápia jelenleg vizsgálatok tárgya (2, 3).
Adatok utalnak arra, hogy a peripartum cardiomyopathia és a dilatatív cardiomyopathia (DCM) esetek egy részében átfedés észlelhető. Erre utal egyrészt az a megfigyelés, hogy a DCM öröklődő formájában, a familiáris DCM által érintett egyes családtagokban PPC kialakulását észlelték, másrészt a PPC által érintett nőbetegek családszűrésénél DCM-es betegeket lehetett igazolni a családtagok között (4–6). Egyes PPC-betegek genetikai szűrése olyan genetikai mutációkat igazolt, amelyek DCM-ben is előfordulnak, mint pl. a béta-miozin (MYH7) és troponin C (TNNC1) géneket érintő mutációk. Egy nemrégi vizsgálat szerint mind a PPC-t, mind a DCM-et okozó genetikai eltérések között a titin (TTN) gén érintettsége a leggyakoribb, amely közös genetikai prediszponáltságra utal (7, 8). Ezen adatok alapján a PPC egyes esetei terhesség alatt manifesztáló, larvált DCM-eseteket jelentenek, és olyan öröklődő, monogénes betegségnek felelnek meg, mint a familáris cardiomyopathiák (9–11), cardiomyopathia fenokópiák (12, 13) vagy a familiáris ioncsatorna-betegségek (14–17).
Közleményünkben egy ilyen peripartum cardiomyopathia esetet mutatunk be, amelynek hátterében genetikai vizsgálattal kettős, titin és desmoplakin génmutációt lehetett kimutatni. Az eset további érdekességét az adja, hogy a válságos klinikai kép sürgős szívtranszplantációt tett szükségessé, amelyet Szegeden végeztek el, mint az első és mind ez idáig egyetlen, vidéken végzett szívtranszplantációt.
Beteg és módszer
Esetismertetés
Az észlelésekor 36 éves, második terhességét viselő nőbeteg terhessége 38. hetében, 2005. 05. 02-án kerül az SBO-ra nehézlégzés miatt, amelynek hátterében az echokardiográfiás vizsgálat tágult bal kamrát, csökkent balkamra-funkciót (EF: 30%) írt le II. fokú mitrális inszufficiencia mellett. 2005. 05. 03-án az Aneszteziológiai és Intenzív Terápiás Intézet Intenzív Osztályára került, ahol nyugalmi dyspnoét, lábszárödémát észleltek, enyhén emelkedett májfunkciós értékek mellett. Az ott készült echokardiográfia súlyosan csökkent balkamra-funkciót (EF: 19%), III-as fokú mitrális inszufficienciát, emelkedett pulmonalis nyomást írt le (PAP: 60 Hgmm), mellkaröntgenen jelentős cardiomegaliával (1. ábra). A vérgáz vizsgálata respiratórikusan kompenzált metabolikus acidózist igazolt. A fentiek alapján peripartum cardiomyopathiát véleményeztünk. Dobutamin, diuretikum mellett az állapota stabilizálható volt. 2005. 05. 04-én sectio caesarea útján egészséges kislánynak adott életet, a műtét közben hemodinamikailag stabil volt. Belgyógyászati ITO-ra való visszavétele után kamrai aritmiák miatt amiodaront kapott, majd 2005. 05. 05-én súlyos szívelégtelenség tünetei miatt levosimendanterápia indult intraaortikus ballonpumpa behelyezése mellett. A következő héten levosimendan, IABP, dopamin, dobutamin mellett állapota hemodinamikailag viszonylag stabil volt, de a terápia csökkentését nem tolerálta. 2005. 05. 12-én kapcsolatfelvétel történt a transzplantációs központtal, 05. 19-én negatív eredményű koronarográfia és szívizom-biopszia történt. 2005. 05. 24-én fokozódó fulladás, hypoxia miatt respirátorkezelés vált szükségessé, a balkamra-diszfunkció tovább súlyosbodott (EF 14%). Jobb szívfél katéterezése alacsony perctérfogat-szindrómát igazolt (CI: 1,4 l/min/m2, SV: 21 ml, SVRI: 3590 dynes/sec/cm–5/m2, PVRI: 58 dynes/sec/cm–5/m2, PAP: 23 Hgmm). A válságos állapot miatt egyedüli megoldásként sürgős szívtranszplantáció jött szóba, amelyet a beteg szállíthatatlan állapota miatt Szegeden végeztek el 2005. 05. 25-én a budapesti Városmajori Ér- és Szívsebészeti Klinika transzplantációs teamje közreműködésével. A perfúziós idő 112 perc, a graft iszkémiás idő 3 óra 16 perc volt.
A beteg családszűrése során derült fény arra, hogy édesanyja dilatatív cardiomyopathiában szenved. Utóbbi alapján familiáris cardiomyopathia merült fel, és genetikai vizsgálat történt a beteg tájékoztatása és beleegyezése alapján.
A vizsgálatot a Szegedi Tudományegyetem Szent-Györgyi Albert Klinikai Központ Regionális Humán Orvosbiológiai Tudományo és Kutatásetikai Bizottsága a 165/2016-SZTE számon engedélyezte.

Genetikai vizsgálat
A genetikai vizsgálat során célzott újraszekvenálással 103, ismert cardiomyopathiát okozó gént vizsgáltunk. Utóbbi az Agilent „SureSelect” technológiát használja egyedi tervezésű, célrégió-specifikus 120 bp. hosszú RNS „bait”-ekkel (Agilent Technologies, Santa Clara, CA, United States). A sokszorosított DNS szekvenálását SOLiD 5500xl Systemmel (Life Technologies, Grand Island, NY, United States) végeztük. A SOLiD readek mappingjét a Genomic Workbench ver 7.0.3-mal (CLC Bio, Qiagen) végeztük, a Human Genome Assembly hg19-et mint referenciaszekvenciát használva. A variánslehívás és variánsannotáció ugyanezzel a szoftverrel készült. A misszensz mutációk által okozott aminosavcserék funkcionális hatását a SIFT és PROVEAN predikciós programokkal elemeztük.
Az azonosított genetikai variánsok validálásra kerültek direkt kapilláris szekvenálás által (BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems), amely során a titin- (TTN) és desmoplakin- (DSP) gének kódoló szakaszainak szekvenálása történt meg ABI Prism 310 Genetic Analyzeren (Applied Biosystems). Az elektroferogramokat a gyártó Sequencing Analyzer v5.4 szoftverével analizáltuk.
Eredmények
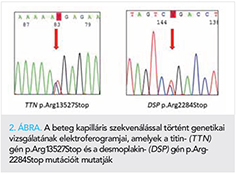
A genetikai vizsgálat során összesen 5 variánst detektáltunk. A variánsok közül kettő, a titingén (TTN) p.Arg13527Stop variánsa (rs374140736); és a desmoplakingén (DSP) p.Arg2284Stop (rs794728130) variánsa bizonyult kóroki variánsnak (2. ábra). Mindkét variáns patogén-besorolással szerepel a ClinVar adatbázisban. További három variáns (TTN p.Glu22041Gln, p.Thr18440Ile; JUP p.Val456Ile) bizonytalan jelentőségű variáns (VUS) megjelöléssel szerepel az adatbázisokban. A feltételezetten kóroki variánsok közül mindkét esetben csonkoló hatású, a fehérje tekintetében funkcióvesztéses mutációt jelentenek.
Megbeszélés
Munkánkban egy kettős, TTN p.Arg13527Stop és DSP p.Arg2284Stop mutációt azonosítottunk súlyos klinikai megjelenésű peripartum cardiomyopathia hátterében, amely sürgős szívtranszplantációt tett szükségessé.
A PPC családi halmozódása ismert jelenség, egy német vizsgálat az általuk vizsgált betegcsoport 15%-ában észlelte a cardiomyopathia valamilyen formájú familiáris megjelenését (6). Családi halmozódást mutató tizennyolc eset közül Spaendonck-Zwarts és munkatársai 4 esetben (22%) találtak patogén genetikai mutációt, amelyek közül három esetben a titingén volt érintett. A 20 PPC-beteg közül mindössze kettőben normalizálódott a bal kamrai ejekciós frakció (7). Egy másik, családi halmozódás szempontjából nem szelektált csoportban 172 PPC-eset genetikai vizsgálatakor 15%-ban találtak patogén genetikai eltérést, amelyek 2/3 része a titingént érintette. A mutációk nagy része a titinfehérjének a sarcomer A sávjában lokalizálódó aminosavjait érintette, hasonlóan a DCM-et okozó titinmutációkhoz. Érdekes módon a titinmutációk mind kaukázusi, mind afroamerikai etnikumban előfordultak, és a titinvariánsok jelenléte összefüggött az 1 éves utánkövetéskor mért alacsonyabb bal kamrai ejekciós frakcióval (8).
Az általunk észlelt mindkét variánst közölték már korábban a szakirodalomban, mint kóroki variánst. A TTN p.Arg13527Stop variánst DCM-ben írták le kóroki variánsként. Norton és munkatársai egy DCM-es család hat családtagjában észlelték, amelyek közül három családtag DCM-ben, egy családtag pedig balkamra-megnagyobbodás nélküli szisztolés diszfunkcióban szenvedett (18). Chanavat és munkatársai egy sporadikus DCM-esetben írták le fenti TTN-mutáció jelenlétét (19). A TTN p.Arg13527Stop-mutáció – a DCM-et okozó TTN-mutációkhoz hasonlóan – a titingén a sarcomer A sávjában lokalizálódó aminosavját érinti. A DSP p.Arg2284Stop variánst aritmogén jobb kamrai cardiomyopathiával (ARVD) és Carvajal-szindrómával (gyapjas haj, palmoplantaris keratoderma és DCM triásza) hozták kapcsolatba (20, 21). Mindkét mutáció funkcióvesztő mutációnak tartható, vagy a stop kodon miatt átíródó csonkolt fehérje keletkezése, vagy a „nonsense mediated mRNA decay” miatt lebomló fehérje miatt kialakuló haploinszufficiencia miatt. Esetünk érdekessége, hogy eddig még nem publikáltak olyan esetet, ahol ez a két patogén variáns együtt jelent volna meg.
Következtetés
Esetünk felhívja a figyelmet arra, hogy a peripartum cardiomyopathia az esetek egy részében genetikai eredetű, ezért a PPC-betegek családszűrése és adott esetben genetikai vizsgálata javasolt.
Köszönetnyilvánítás
A szerzők köszönetüket fejezik ki prof. dr. Szabolcs Zoltán profilvezető szívsebésznek és a Városmajori Szív- és Érgyógyászati Klinika általa vezetett szívtranszplantációs munkacsoportjának a szívtranszplantációban nyújtott önzetlen szakmai segítségükért és elévülhetetlen érdemeikért. A munka a „Ritka betegségek patogenezisének kutatása, új diagnosztikai és terápiás eljárásokat megalapozó fejlesztések” (GINOP-2.3.2-15-2016-00039), az „Életet veSzélyezTető Akut megbetegedések súlYossági és hALálozási mutatóinak jaVítása transzlációs orvostudományi mEgközelítésben – STAY ALIVE” (GINOP-2.3.2-15-2016-00048) és a Szegedi Tudományegyetem ÁOK Kari Kutatási Alap „Hetényi Géza” pályázatának támogatásával készült.
Irodalom
1. Sliwa K, Hilfiker-Kleiner D, Petrie MC, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. European Journal of Heart Failure 2010; 12: 767–778. https://doi.org/10.1093/eurjhf/hfq120
2. Honigberg MC, Givertz MM. Peripartum cardiomyopathy. BMJ 2019; 364:k5287. https://doi.org/10.1136/bmj.k5287
3. Arany Z, Elkayam U. Peripartum Cardiomyopathy. Circulation 2016; 133: 1397–1409. https://doi.org/10.1161/CIRCULATIONAHA.115.020491.
4. Pearl W. Familial occurrence of peripartum cardiomyopathy. Am Heart J 1995; 129: 421–422.
5. Pierce JA, Price BO, Joyce JW. Familial occurrence of postpartal heart failure. Arch Intern Med 1963; 111: 651–655.
6. Haghikia A, Podewski E, Libhaber E, et al. Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res Cardiol 2013; 108: 366. https://doi.org/10.1007/s00395-013-0366-9.
7. van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 2014; 35: 2165–2173. https://doi.org/10.1093/eurheartj/ehu050.
8. Ware JS, Li J, Mazaika E, Yasso CM, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016; 374: 233–241. https://doi.org/10.1056/NEJMoa1505517.
9. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733–2779. https://doi.org/10.1093/eurheartj/ehu284.
10. Toth T, Nagy V, Faludi R, et al. The Gln1233ter mutation of the myosin binding protein C gene: Causative mutation or innocent polymorphism in patients with hypertrophic cardiomyopathy? Int J Cardiol 2011; 153(2): 216–9. https://doi.org/10.1016/j.ijcard.2011.09.062.
11. Orosz A, Baczko I, Nagy V, et al. Short-term beat-to-beat variability of the QT interval is increased and correlates with parameters of left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Can J Physiol Pharmacol 2015; 93(9): 765–72. https://doi.org/10.1139/cjpp-2014-0526.
12. Csanyi B, Popoiu A, Hategan L, et al. Identification of two novel LAMP2 gene mutations in Danon disease. Can J Cardiol 2016; 32(11): 1355.e23–1355.e30. https://doi.org/10.1016/j.cjca.2016.02.071.
13. Csányi B, Hategan L, Nagy V, et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease with a Predominant Cardiac Phenotype. Int Heart J 2017; 58(3): 454–458. https://doi.org/10.1536/ihj.16-361
14. Csanády M, Kiss Z. Az elektrokardiogram QT-távolságának örökletes megnyúltsága, veleszületett süketség nélkül (Romano-Ward-syndroma). Orv Hetil 1972; 47: 2840–2843.
15. Sepp R, Hategan L, Bácsi A, et al. Timothy Syndrome 1 Genotype without Syndactyly and Major Extracardiac Manifestations. Am J Med Genet A 2017; 173(3): 784–789. https://doi.org/10.1002/ajmg.a.38084.
16. Hategan L, Csányi B, Ördög B, et al. A novel ‘splice site’ HCN4 gene mutation, c.1737+1 G>T, causes familial bradycardia, reduced heart rate response, impaired chronotropic competence and increased short-term heart rate variability. Int J Cardiol 2017; 241: 364–372. https://doi.org/10.1016/j.ijcard.2017.04.058.
17. Ördög B, Hategan L, Kovács M, et al. Identification and functional characterisation of a novel KCNJ2 mutation, Val302del, causing Andersen-Tawil syndrome. Can J Physiol Pharmacol 2015; 93(7): 569–75. https://doi.org/10.1139/cjpp-2014-0527.
18. Norton N, Li D, Rampersaud E, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet 2013; 6: 144–53. https://doi.org/10.1161/CIRCGENETICS.111.000062.
19. Chanavat V, Janin A, Millat G. A fast and cost-effective molecular diagnostic tool for genetic diseases involved in sudden cardiac death. Clin Chim Acta. 2016; 453: 80–5. https://doi.org/10.1016/j.cca.2015.12.011.
20 Fressart V, Duthoit G, Donal E, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 2010; 12: 861–8. https://doi.org/10.1093/europace/euq104.
21. Antonov NK, Kingsbery MY, Rohena LO, et al. Early-onset heart failure, alopecia, and cutaneous abnormalities associated with a novel compound heterozygous mutation in desmoplakin. Pediatr Dermatol 2015; 32: 102–8. https://doi.org/10.1111/pde.12484.