Genetic testing in hereditary cardiovascular diseases
█ Review
DOI: 10.26430/CHUNGARICA.2022.52.5.23
Authors:
Nagy Beáta1, Csonka Katalin1, Fekete Bálint András, Dohy Zsófia2,
Szabó Liliána2, Fintha Attila1, Matolcsy András1, Merkely Béla2, 3,
Vágó Hajnalka2,3*, Bödör Csaba1,4*
1Semmelweis Egyetem, Patológiai és Kísérleti Rákkutató Intézet, Budapest
2Semmelweis Egyetem, Városmajori Szív- és Érgyógyászati Klinika, Budapest
3Semmelweis Egyetem, Sportorvostan Tanszék, Budapest
4Semmelweis Egyetem, HCEMM-SE Molekuláris Onkohematológia Kutatócsoport, Patológiai és Kísérleti
Rákkutató Intézet, Budapest
Levelezési cím:
Dr. Vágó Hajnalka, e-mail: vago.hajnalka@med.semmelweis-univ.hu,
Dr. Bödör Csaba, e-mail: bodor.csaba1@med.semmelweis-univ.hu
*Egyenlően járultak hozzá.
Summary
Our knowledge in cardiovascular diseases, such as cardiomyopathies, arrhythmias and aortopathies has expanded in the last few years owing to the dynamic development of DNA-sequencing methods. Besides better understanding the genetic background of these diseases, progression has been made in therapeutic arena of these diseases as well, requiring precise diagnosis and advanced patient risk stratification. Genetic testing as a diagnostic tool helps to refine the prognosis of the disease and paves the way towards a personalized patient management. The aim of genetic testing is to identify the disease-causing variants in the index patient, followed by presymptomatic or predictive testing of the relatives with increased risk for the disease. Cardiologists play a key role in selection of patients who may benefit from genetic testing, to which understanding of basics of genetic analyses is necessary. This article serves as a short overview of the genetic background of common hereditary cardiovascular diseases, general rules of clinical genetics and practical application of cardiogenetic testing. Finally, we present our initial genetic testing results in this patient group.
ISSUE: CARDIOLOGIA HUNGARICA | 2022 | VOLUME 52, ISSUE 1
Összefoglalás
A DNS-szekvenálási technológiákban bekövetkezett dinamikus fejlődésnek köszönhetően ismereteink számos kardiológiai betegség, így a cardiomyopathiák, az aritmiaszindrómák és az aortopathiák genetikai alapjainak megértésében is jelentősen bővültek az elmúlt években. A genetikai háttér jobb megértése mellett előrelépések történtek a kezelési lehetőségek terén is, amelyek megkövetelik a pontos diagnózisalkotást és a páciensek rizikóbecslését. A genetikai vizsgálatok integrációja a diagnosztikai eljárások sorába segít a prognózis meghatározásában és a személyre szabott klinikai menedzsment kialakításában. A genetikai vizsgálat célja a betegséget okozó mutáció azonosítása, majd a fokozott rizikójú családtagok preszimptomatikus vagy prediktív tesztelése. A kardiológus kulcsszerepet tölt be azon páciensek kiválasztásában, akik profitálhatnak a genetikai vizsgálatból, amihez elengedhetetlen a genetikai vizsgálatok alapjainak ismerete. Jelen összefoglaló közleményben rövid áttekintést adunk a gyakoribb örökletes kardiovaszkuláris betegségek genetikai hátteréről, a klinikai genetika általános szabályairól, a kardiogenetikai vizsgálat gyakorlati alkalmazásáról és bemutatjuk eddigi vizsgálati eredményeinket ezen betegcsoportokban.
Bevezetés
A humán betegségek patomechanizmusának megértésében hatalmas előrelépésnek számított az emberi genom szekvenciájának feltérképezése a kétezres évek elején (1). Néhány évvel később az új generációs szekvenálás (NGS) bevezetésével a technológia költségei drámaian csökkentek, így széles körben elérhetővé vált a kutatólaboratóriumok számára, amellyel a legtöbb betegség genetikai hátterének megismerése lehetővé vált. Azonban továbbra is számos gén biológiai jelentősége tisztázatlan, és egy betegséget okozó genetikai eltérés több fenotípus képében is megjelenhet, amely további módosító tényezők, környezeti hatások szerepére utal, befolyásolva ezzel a kiváltó genetikai eltérés expresszióját. Az új technológiáknak köszönhetően a kardiológiai betegségek területén is bővült a tudásunk, így a diagnosztikában, családszűrésben, rizikóstratifikációban is precíziós orvoslás irányú szemléletváltás történt (2). Napjainkban a genetikai vizsgálatok a kardiológiai klinikai gyakorlat részévé váltak, így a Semmelweis Egyetemen is beállításra került egy 174, kardiovaszkuláris betegségekhez kapcsolódó génből álló NGS-vizsgálat a magyar kardiológiai betegek teljes körű kivizsgálásának céljából. Az elmúlt egy évben közel 100 beteg kardiogenetikai vizsgálatát végeztük el, akik klinikai genetikai tanácsadás keretében vehették át elkészült leleteiket.
Genetikai vizsgálatok szerepe az egyes betegségcsoportokban
Cardiomyopathiák
A cardiomyopathia hátterében gyakran áll patogén genetikai eltérés, beleértve a hipertrófiás cardiomiopathiát (HCM), a dilatatív cardiomyopathiát (DCM), aritmogén cardiomyopathiát (ACM), a restriktív cardiomyopathiát (RVC), és a bal kamrai non-kompakt cardiomyopathiát (LVNC, left ventricular non-compaction cardiomyopathy). A cardiomyopathia hátterében álló leggyakoribb géneket és a szívizomszövetben betöltött szerepüket az 1. táblázat tartalmazza.
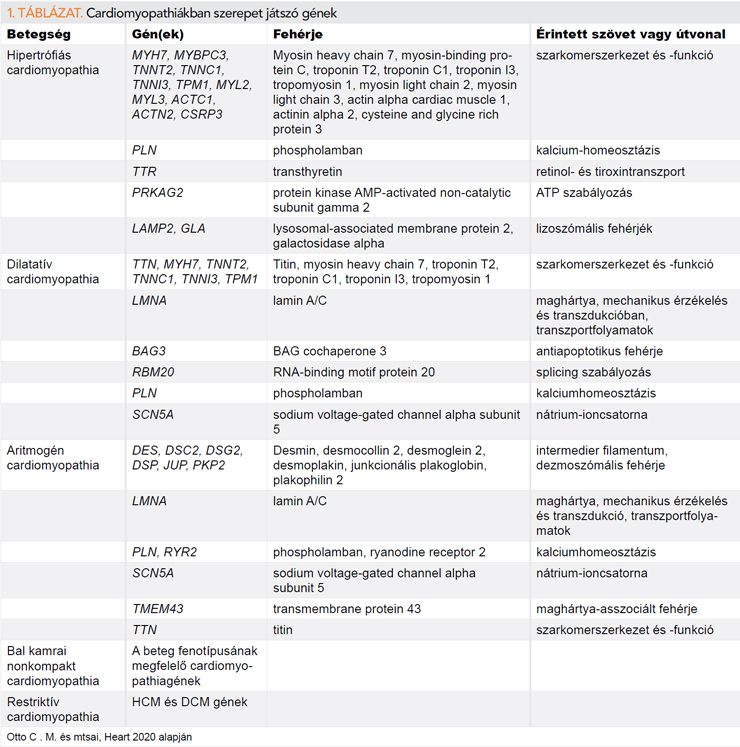
Hipertrófiás cardiomyopathia
A genetikai vizsgálat fontos szerepet tölt be a HCM diagnózisában, valamint a betegek és családtagjainak kivizsgálásában és kezelésében. A HCM autoszomális domináns öröklődésű, így az utódokban 50% eséllyel jelenik meg ugyanaz a betegséget okozó genetikai variáns, mint a szülőben (3). Genetikai konzultáció és genetikai vizsgálat elvégzése ajánlott minden HCM-es fenotípusú beteg esetén, amely megerősítheti a diagnózist és lehetővé teszi a veszélyeztetett családtagok szűrését is (4). A HCM-et elsősorban a szarkomer proteineket kódoló gének mutációi okozzák, amely gének rosszul tolerálják a genetikai variabilitást. Génpanelünk tartalmazza a HCM-ben vizsgált 8 leggyakoribb szarkomer gént: ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNI3, TNNT2, TPM1. A szarkomer géneken túl fontos a betegekben a GAA (Pompe-kór), PRKAG2 (glikogéntárolási-betegség), LAMP2 (Danon-betegség) és GLA (Fabry-kór) fenokópia géneket is analizálni, amelyek klinikailag hipertrófiás cardiomyopathia képében jelenhetnek meg. Fabry-kór esetében az α-galaktozidáz-A enzimaktivitás mérése az ismeretlen szignifikanciájú GLA-variánsok besorolásában is segíthet (5).
Dilatatív cardiomyopathia
A DCM-re kifejezett lókusz-heterogenitás jellemző, a többi cardiomyopathiával ellentétben patogenezisében számos genetikai eltérés szerepet játszhat (6). Többek között citoszkeletális (DES), mitokondriális (tRNS), szarkomerikus (TTN, TNNI3, TNNT2, TPM1, MYBPC3, MYH7, FLNC), dezmoszomális (DSP), maghártya-asszociált (LMNA) és RNS-kötő fehérjéket kódoló (RBM20) gének hozhatók összefüggésbe a DCM kialakulásával. A legismertebb közülük a TTN-gén, amely a legnagyobb szívben expresszálódó proteint, a titint kódolja. A titin a szarkomer kontrakcióját és jelátvitelét szabályozza. A TTN a szív fejlődése során alternatív splicing segítségével igazodik a fiziológiai állapothoz, amely megnehezíti a genetikai analízisét. A TTN missense mutációi mellett egyre nagyobb jelentőséget tulajdonítanak a trunkáló variánsoknak, amelyek megváltoztatják a titin szerkezetét, és az esetek 18-25%-ában felelősek a DCM kialakulásáért (7). Jelen tudásunk szerint trunkáló variánst hordozó egyéneknél részletes kardiológiai klinikai kivizsgálás szükséges, azonban, feltehetően az inkomplett penetrancia miatt, összefüggése a betegség prognózisával nem egyértelmű (8).
A lamin A/C-gén (LMNA) mutációi a második leggyakrabban azonosított eltérések DCM-ben. Az LMNA egy intermedier filamentum proteint kódol, amely szerepet játszik a sejtmag strukturális integritásának fenntartásában, génexpressziós szabályozásban, mechanikus érzékelésben és transzdukcióban, valamint transzportfolyamatokban. Az LMNA-mutációk gyakran ingerületvezetési zavarokat, kamrai aritmiákat vagy akár hirtelen szívhalált is okozhatnak, amelynek patomechanizmusa ma is aktívan kutatott terület. Az LMNA-mutáció típusa befolyásolhatja a cardiomyopathia prognózisát és terápiáját is, mivel a non-missense (indel/trunkáló, splicing régiót érintő) LMNA-variánsok magasabb aritmiarizikóval járhatnak, mint a missense mutációk (9). Az American College of Cardiology (ACC)/American Heart Association (AHA)/Heart Rhythm Society (HRS) 2017-es ajánlása szerint azon Lamin A/C-mutáció okozta cardiomyopathiában szenvedő betegek esetében, akik legalább két rizikófaktorral (nem tartós kamrai tachycardia, LVEF <45%, non-missense mutáció, férfinem) bírnak, a hirtelen szívhalál megelőzése céljából primer prevenciós implantálható kardioverter-defibrillátor (ICD) beültetése ajánlott (IIa osztály, B-evidenciaszint) (10). Az Európai Kardiológusok Társasága 2020-ban megjelent Sportkardiológiai ajánlása alapján is az LMNA/C-mutációval rendelkező sportolók szigorúbb megítélése szükséges, náluk a nagy intenzitású sporttevékenység nem ajánlott függetlenül a klinikai panaszok, illetve bal kamrai ejekciós frakciótól (11). Az LMNA-asszociált DCM forrongó terület gyógyszerkutatás szempontjából is. Ezen mutációt hordozó betegekben az mTOR útvonal és a MAPK-jelátvitel aktiválódását figyelték meg, amelyek esetleges terápiás célponttá válhatnak a jövőben (9).
Aritmogén cardiomyopathia
Az aritmogén jobb kamrai cardiomyopathia (ARVC) genetikai és fenotípusbeli átfedést mutat az aritmogén bal kamrai cardiomyopathiával, illetve sok esetben kétkamrás érintettség van jelen, ezért manapság összefoglaló néven aritmogén cardiomyopathiaként (ACM) emlegetik ezt az entitást. Az ACM-et kezdetben a dezmoszóma betegségének tartották, mivel az ACM-esetek több mint felében a DSC2, DSG2, DSP, PKP2, JUP dezmoszomális fehérjéket kódoló gének mutációi azonosíthatóak (1. táblázat) (12). A genetikai elemzést nehezítheti a digénes vagy poligénes öröklődés és összetett („compound”) heterozigótaság, ami emellett változó expresszivitással vagy csökkent penetranciával társulhat. Nagy intenzitású sporttevékenység az ACM-es betegeknél egyértelműen a betegség progresszióját eredményezi, így a jobbkamra-tágulat kifejezettebbé válását, és a malignus kamrai ritmuszavarok gyakoribb előfordulását. Emiatt az ESC Sportkardiológiai ajánlása alapján az intenzív sporttevékenység végzése nem javasolt ezen betegeknél, beleértve a fenotípus negatív, de genetikailag igazolt dezmoszómális génvariánst (11). A strukturális átrendeződés mértéke kulcsfontosságú a kamrai aritmiák keletkezésében. A dezmoszómális gének mutációi nem specifikusak ACM-re, ezen patogén variánsait DCM-es betegek vizsgálata során is leírták (13). Multigénes panelekkel végzett elemzések során fény derült további, nem dezmoszómális gének (LMNA, RBM20, FLNC) szerepére is az ACM patogenezisében.
Restriktív cardiomyopathia
Az RCM ritka cardiomyopathia, amely lehet örökletes vagy szerzett, de szisztémás betegség részeként is megjelenhet. Az idiopátiás RCM-esetek több mint 60%-ában található patogén vagy valószínűleg patogén mutáció, többek között a MYH7, DES, FLNC, MYBPC3, LMNA génekben, amely alapján ezen betegcsoportban genetikai vizsgálat elvégzése indokolt (14). A szívamiloidózis különböző típusait fontos elkülöníteni az egyéb RCM-formáktól a betegség progresszív lefolyása és a különböző kezelési stratégiák miatt. Az elmúlt évek kutatásainak eredményeként jelentős fejlődésnek lehettünk szemtanúi az örökletes transztiretin amiloidózis célzott terápiáját illetően, amely ösztönzően hat a genetikai vizsgálatokra is (15). Az RCM hátterében ritkán állhat hemokromatózis is, amely a szérumvas, ferritin és transzferrin szaturációjának mérésével könnyen diagnosztizálható. A hemokromatózis leggyakoribb formájában a HFE-gén homozigóta mutációi igazolhatók, fontos megjegyezni azonban, hogy a kaukázusi populáció 6%-a heterozigóta és 0,4%-a homozigóta a HFE mutációira nézve, amelyek nem feltétlenül jelentenek fokozott rizikót a megbetegedésre (16).
Bal kamrai nonkompakt cardiomyopathia
Az LVNC három anatómiai eltérésből tevődik össze: kifejezett bal kamrai trabekuláltság, vékony kompaktizomzat, és mély intertrabekuláris recesszusok. Non-kompaktáció megfigyelhető egészséges egyénekben, sportolókban, terhes nőkben vagy szisztémás betegségekhez társulóan is, ezért az LVNC-cardiomyopathia diagnózisa megfelelő körültekintéssel állítható csak fel, jó balkamra-funkció, negatív személyes és családi anamnézis mellett inkább morfológiai leírásként alkalmazandó. Mivel LVNC előfordulhat DCM, HCM, RCM, ACM fenotípusokkal együtt, ezért a genetikai vizsgálat során talált variánsok inkább a háttérben álló cardiomyopathia genetikai okaiként értelmezhetők, és nem a non-kompaktációt kiváltó mutációként (17). Klinikai családszűrés elvégzése javasolt annak feltérképezésére, hogy a non-kompakt jellemvonás örökletes vagy sporadikus. A jelenlegi ajánlások szerint genetikai vizsgálat ajánlott cardiomyopathia és LVNC együttes előfordulása, LVNC-vel társuló szindrómák, vagy véletlenül felfedezett LVNC esetén (18). Jelenleg nem ismert LVNC-vel egyértelműen összefüggésbe hozható genetikai eltérés, ezért rutinszerűen a cardiomyopathiára jellemző gének vizsgálata javasolt (19). Tünetmentes, egyébként normál szívfunkciójú egyénben azonosított LVNC-fenotípus esetén genetikai analízis nem szükséges.
Vaszkuláris megbetegedések/aortopathiák
Az ereket leggyakrabban érintő betegség az ateroszklerózis, amelynek genetikai háttere komplex, patogenezisében több száz vagy ezer egy nukleotid polimorfizmus (single nucleotid polymorphism, SNP) játszhat szerepet, amelyek egyenként kis hatással bírnak a betegség kialakulására. Azonban léteznek olyan érbetegségek is, amelyek létrejöttéhez egyetlen gén eltérése elegendő, ezek jellemzően Mendeli-öröklésmenetet mutatnak. A mellkasi aortaaneurizmával járó betegségek a leggyakoribbak a monogénes, artériadilatációt okozó kórképek közül. Közel 30%-ukhoz köthető patogén genetikai eltérés, döntően a simaizomsejt-funkcióban szerepet játszó proteineket, extracelluláris mátrix integritását fenntartó fehérjéket kódoló génekben és a TGF-ß jelátviteli útvonalban. A Marfan- (MFS), Loeys–Dietz- (LDS) és vaszkuláris Ehlers–Danlos- (vEDS) szindrómák tünetei átfedőek lehetnek, így gyakran genetikai vizsgálat szükséges a végleges diagnózis felállításához. Nem szindromatikus esetekben általában hiányoznak a nonkardiovaszkuláris eltérések, ezért nincsenek nyilvánvaló fizikai jellegzetességeik, így a genotípus meghatározása segíthet a pontos diagnózis felállításában és a betegség prognózisának megállapításában is. Szinte minden LDS vagy vEDS esetén található patogén vagy valószínűleg patogén variáns a társuló génekben (20, 21). Ismertek specifikus genotípus-fenotípus összefüggések is egyes betegségekben, amelyek irányíthatják a gyógyszeres vagy sebészi kezelést, javítva ezzel a betegség kimenetelét. A vEDS kialakulásáért felelős COL3A1-génben kialakuló glicinszubsztitúció, inframe inzerció vagy deléció kevésbé súlyos fenotípussal társul, mint a haploinszufficienciához vezető nullallélok létrejötte (22). Az MFS-es betegekben a ciszteinmaradványokat érintő FBN1-variánsok esetén nagyobb valószínűséggel alakul ki aortadilatáció és szemészeti manifesztáció, míg a trunkáló vagy splicing FBN1-variánst hordozó betegekben gyakrabban alakul ki aortadisszekció és nagyobb valószínűséggel szorulnak műtétre. Az FBN1-gén 24-32-es exonjainak mutációi esetén a betegség már fiatalabb korban manifesztálódik, és súlyos aortakomplikációkkal társul (23). A Clinical Genome Resource (ClinGen) ajánlása szerint 9 gén társítható egyértelműen örökletes mellkasi aortaaneurizmával és disszekcióval járó betegségekhez: ACTA2, COL3A1, FBN1, MYH11, MYLK, SMAD3, TGF-ß2, TGF-ßR1, és TGF-ßR2 (24).
Aritmiák
A hosszú QT-szindróma (Long QT syndrome, LQTS) és a Brugada-szindróma (BrS) potenciálisan letális kimenetelű kamrai ritmuszavarokkal járó betegségek, ezért az elmúlt években intenzív kutatások folytak a diagnosztikájuk és kezelési lehetőségeik javítása érdekében.
A veleszületett LQTS a szoros genotípus-fenotípus összefüggéseknek köszönhetően egyike azon kevés monogénes kardiovaszkuláris betegségnek, amelyben a genetikai eltérés diagnosztikai szereppel bír, meghatározza a prognózist és befolyásolhatja a terápiás döntést is. Az American College of Cardiology (ACC)/ American Heart Association (AHA) és a European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS) szerint I. osztályú ajánlás genetikai tanácsadást és genetikai vizsgálatot végezni a következő esetekben:
- erős klinikai gyanú esetén (Schwartz-score ≥3,5);
- tünetmentes, negatív családi anamnézisű esetekben, ha a sorozat EKG-n idiopátiás QTc-megnyúlás látható; és
- rokonok kaszkád szűrése esetén, ha az index páciensnél patogén/valószínűleg patogén mutációt azonosítottak.
Klinikailag hosszú QT-szindróma gyanús betegek 80%-ában található mutáció a 3 kanonikus, LQTS-ra hajlamosító génben (KCNQ1/LQT1, KCNH2/LQT2 és SCN5A/LQTS3) (25, 26). A genotípus-fenotípus összefüggések szerint LQTS1-ben az emocionális stressz és fizikai aktivitás, míg LQTS2-ben a hirtelen zaj, hypokalaemia és szülés utáni időszak lehet triggere a kardiális eseményeknek, LQTS3-ban pedig alvás vagy pihenés közben alakul ki aritmia. A génspecifikus triggerek kerülése LQTS-betegek esetében fontos szereppel bírhat (27).
A LQTS-sel ellentétben a genetikai vizsgálat szerepe a Brugada-szindrómás betegek diagnózisában és klinikai vezetésében napjainkban még jóval kisebb. A BrS hátterében mintegy 20-30%-ban azonosítható a SCN5A patogén vagy valószínűleg patogén funkcióvesztő mutációja. A SCN5A (nátrium-ioncsatornát kódoló gén) az egyetlen hajlamosító gén, amely a ClinGen osztályozás szerint egyértelműen összefüggésbe hozható a BrS-sel (28). A fennmaradó esetekben „minor” hajlamosító gének (CACNA1C, CACNA2D1, CACNB2, PKP2, SCN1B, SCN2B, SCN3B) játszhatnak szerepet, amelyek befolyásolják a nátriumáramlást. Úgy tűnik, a BrS-komplex folyamatok eredménye, amelyben közrejátszik a csökkent depolarizációs tartalékkal járó genetikai hajlam, a kor (29), a férfinem (29), a láz (30), és egyes gyógyszerek is. A BrS patogenezisében feltehetően a genetikai eltérés miatt kialakult nátriumhomeosztázis-zavar játszik kulcsszerepet, amely fokozott miocitanekrózishoz, gyulladáshoz és fibrózishoz vezethet a jobb kamrai kiáramlási traktusban (RVOT) szubepikardiálisan. Ezen területen megszakad az ingerület haladásának folytonossága, amely megteremti az alapját a malignus kamrai ritmuszavarok kialakulásának (31). Az AHA/ACC (32), ESC (33), HRS/EHRA (34) ajánlások alapján a SCN5A-gén specifikus vizsgálata javasolt a klinikailag gyanús páciensben, az esetlegesen érintett elsőfokú rokonok felderítésének céljából.
Már 20 éve annak, hogy leírták az első génmutációt katekolaminerg polimorf ventrikuláris tachycardiában (CPVT) (35). A genetikai vizsgálat alapvető szerepet tölt be a betegség diagnosztikájában, a jelenlegi ajánlások szerint a RYR2- vagy CASQ2-gének patogén variánsai még klinikai fenotípus hiányában is elegendőek a CPVT diagnózisának felállításához (33). A RYR2 (ryanodinreceptor-2) a szarkoplazmás retikulum Ca2+-csatornáját kódolja, amelynek elégtelen működése mutációtól függően különböző mechanizmusokon keresztül, kórosan megnövekedett intracelluláris Ca2+-koncentrációhoz és potenciálisan életveszélyes kamrai ritmuszavarokhoz vezethet. Az autoszomális domináns öröklődésű CPVT-ben azonosított RYR2-variánsok után a CPVT recesszív formájában (CPVT-2) a CASQ2-homozigóta mutációinak szerepét is leírták (36). A CASQ2 a kardiális kalszekvesztrint kódolja, amely a RYR2 makromolekula komplex része, és gátolja a RYR2 nyitását alacsony intraszarkoplazmatikus Ca2+-koncentráció esetén (37). Atípusos CPVT-csoportba sorolhatók azon betegek, akik fenotípusa átfedést mutat a QT-megnyúlással járó eltérésekkel és a katekolamin-mediált kamrai tachycardiákkal. Ezen csoportba tartozó betegeknél a CALM1, CALM2, CALM3 kalmodulint kódoló gének, TRDN kardiális triadint kódoló gén, TECRL transzenoil-CoA reduktáz-szerű fehérjét kódoló gének mutációi játszhatnak szerepet. A RYR2 funkcióvesztő mutációi is ebbe a csoportba sorolhatók. Ezen specifikus genetikai eltérések felfedezése új, célzott, génterápia alapú kezelési perspektívát nyitott CPVT-ben, amely aktívan kutatott terület napjainkban (38).
Kinek javasolt kardiogenetikai vizsgálat?
Kardiogenetikai kivizsgálást az érintett család azon tagjában érdemes kezdeni, akiben a legnagyobb valószínűséggel megtalálható a kóroki variáns. Ez általában a súlyosabb tünetekkel jelentkező vagy a fiatalabb életkorban tünetessé váló egyént jelenti (ún. indexpáciens). Örökletes kardiovaszkuláris betegség gyanúját keltő tüneteket a 2. táblázatban foglaltuk össze. Ha az indexeset autopszia során kerül felfedezésre, posztmortem szövet analízise ajánlott („molekuláris autopszia”). Amennyiben genetikai vizsgálattal olyan valószínűleg patogén/patogén variáns mutatható ki, amely illeszkedik a klinikai jellemzőkhöz, a többi családtag klinikai és genetikai vizsgálata is javasolt. Ha a szülők negatívak az adott variánsra nézve, valószínűleg de novo mutáció alakult ki az indexpáciensben.

Autoszomális domináns öröklődésű cardiomyopathia esetén a páciens utódai 50% eséllyel öröklik a patogén allélt. A genotípus pozitív családtagoknak rendszeres klinikai kontroll javasolt, az adott variánsra negatív testvér vagy gyermek azonban mentesül a további vizsgálatok alól, bennük nem emelkedett a rizikó az adott betegség kialakulására, és tovább sem adhatják utódjaiknak a betegséget.
Mi várható a kardiogenetikai vizsgálattól?
A genetikai eredmények jelentősége egyre nő a kardiológiában, és mára a betegek és családtagjaik széles körű kivizsgálásának részét képezi. A specifikus patogén variáns segíthet a diagnózis felállításában és a prognózis meghatározásában, ismerete befolyásolhatja a beteg vezetését, beleértve a kontrollok gyakoriságát, a megfelelő gyógyszeres kezelést (például enzimpótlás Fabry-kórban), eszközös terápiát (implantálható kardioverter-defibrillátor, ICD), fizikai aktivitást, aortaműtétet vagy szívtranszplantációt. Fabry-kóron kívül a genotípus egyéb területeken is hatással lehet a terápiás döntésre. Cardiomyopathiában az LMNA, FLNC, DSP-gének egyes variánsai súlyosabb aritmiákkal társulnak, emiatt ezen esetekben korábbi antiaritmiás kezelés szükséges (32, 39, 40). A fent említett okok mellett a kardiogenetikai vizsgálat egyik legfontosabb célja, hogy akár tünetmentes állapotban felfedje az adott betegségre magas rizikóval bíró családtagokat, és emiatt költséghatékonyabb vizsgálat, mint önállóan a klinikai szűrés (41). Fiatal betegek esetében családtervezés szempontjából is kiemelt jelentőségű a gyermek genetikai rizikójának meghatározása.
Az új generációs szekvenálás (NGS) szerepe az örökletes kardiovaszkuláris megbetegedésekben (rövid technológiai összefoglalás)
Az új generációs szekvenálás (Next Generation Sequencing, NGS) lehetővé teszi bármely kardiovaszkuláris fenotípushoz kapcsolódó gének egyidejű tesztelését, így napjainkra a multigénes molekuláris genetikai vizsgálatok a gyakorlatban is irányadó módszerré váltak a szív- és érrendszeri betegségek gyógyászatában (42). Az NGS sikeresnek bizonyult az új, kóroki mutációk azonosításában és a Mendeli-betegségek diagnosztikájában, amelyek akár egyetlen gén egyetlen variánsára vezethetők vissza. Ez a technológia lehetőséget biztosít több gén párhuzamos elemzésére is anélkül, hogy ismernénk az egyes betegségek hátterében álló biológiai mechanizmusokat, ezzel hozzájárulva ismereteink bővítéséhez a komplex betegségek patológiájával kapcsolatban. Továbbá, az NGS hasznos módszer lehet a ritka variánsok azonosítására kisebb családokban is, így lehetőség van az egyénre szabott és informatív tanácsadásra a családtagok számára (43).
Az NGS kifejezés több szekvenálási technológia leírására használatos, amelyek lehetővé teszik több DNS- és RNS-molekula egyidejű szekvenálását. A technikai részletekben mutatkozó különbségek ellenére valamennyi NGS-technológia közös tulajdonságokkal rendelkezik. Például, mindegyiknek szüksége van könyvtár-előkészítési, szekvenálási, képalkotási és adatelemzési folyamatokra. Az NGS technikai részleteit korábbi cikkek részletesen tárgyalják (43–45), így cikkünkben csak rövid áttekintést nyújtunk a szekvenálási munkafolyamat lépéseiről.
Az NGS-rendszer közös lépései a genomi DNS véletlenszerű fragmentálása, majd pedig a könyvtárkészítés, ami lehetővé teszi a nagyszámú gén párhuzamos szekvenálását. Az egyes könyvtárfragmenseket vagy klonálisan amplifikálják emulziós PCR-rel (Roche and Life Technologies) vagy szilárd felületi híd-amplifikációval (Illumina) végzik el, illetve a harmadik generációs (PacBio és MinION) módszert használva egyetlen DNS-molekulát szekvenálnak anélkül, hogy amplifikációra lenne szükség. A minták szekvenálása egy a könyvtár adapterekre komplementer oligonukleotidokkal borított üveglemezen („flow cell”-en) történik fluoreszcens, lumineszcens vagy protonjeleket létrehozva, amelyek detektálható képeket generálnak vagy a pH-detektorok érzékelik azokat. Végül, a kapott jeleket szekvenciarészletekké („read”) alakítják és speciális bioinformatikai analízis segítségével a referenciaszekvenciához illesztik, amit a variánsok azonosítása és annotálása követ (43).
Az NGS-technológiát a fentebb ismertetett kardiovaszkuláris kórképek genetikai profiljának meghatározására a szegedi kardiogenetika munkacsoport alkalmazta először hazánkban. A kutatócsoport két évtizedes munkájának köszönhetően többek közt a HCM (46, 47), DCM (48), hosszú-QT-szindróma (49, 50) betegségekben is történtek új felfedezések.
Az eredmények interpretálása (kategóriák: patogén, valószínűleg patogén, ismeretlen jelentőségű variáns, valószínűleg benignus, benignus)
Napjainkban a genetikai teszteket végző laboratóriumok kiemelt törekvése, hogy az azonosított variánsok interpretációját minél összehangoltabban végezzék el. Az American College of Medical Genetics and Genomics (ACMG) és az Association of Molecular Pathology (AMP) által, 2015-ben közölt irányelvek biztosítják a genetikai leletek, laboratóriumok közötti egységességét és szabványosságát (51).
Jelenleg a variánsokat az ACMG/AMP javaslata alapján 5 fő kategóriába sorolják: patogén („pathogenic”), valószínűleg patogén („likely pathogenic”), ismeretlen jelentőségű („uncertain significance”), valószínűleg benignus („likely benign”), benignus („benign”) (52, 51). Ezen kategóriák definícióját a 3. táblázatban foglaltuk össze.

A klinikumban a variánsok kétféleképpen értelmezhetőek (1. ábra). A patogén és a valószínűleg patogén variánsok pozitív eredménynek tekinthetőek; vagyis a variánsok a betegség okai közé sorolhatóak, illetve a benignus és a valószínűleg benignus variánsok negatív eredménynek tekintendők. A patogén és valószínűleg patogén variánsok megerősíthetik a gyanús betegségek diagnózisát, vagy indokolhatják a klinikai kezelés megváltoztatását. Mindemellett egy patogén vagy valószínűleg patogén osztályozású variáns azonosítása a családtagok genetikai vizsgálatának elvégzését is indokolja (53).
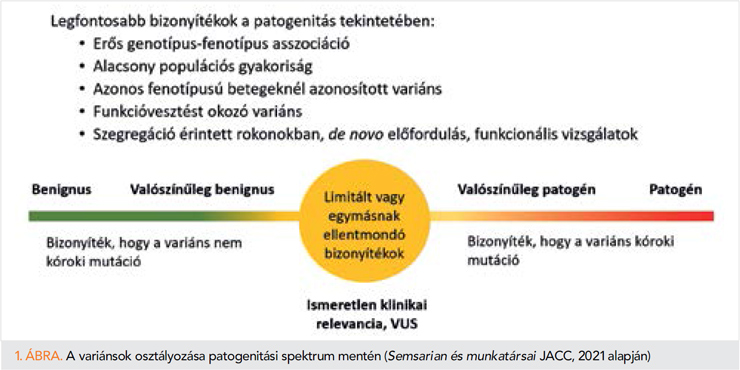
Az ismeretlen jelentőségű variánsok kihívást jelenthetnek a genetikai tesztek eredményeinek értelmezésében, mivel azokat nem tekintik végérvényesen patogénnek, sem pedig végérvényesen benignusnak. Bizonyos esetekben jobb vagy rosszabb prognózisra utalhatnak, azonban egy VUS lehetséges klinikai hatásai ismeretlenek, így a jelenléte nem befolyásolhatja a klinikai döntéshozatalt és nem indokolja közvetlenül a családtagok prediktív célú tesztelését. Fontos megjegyezni, hogy a tesztlaboratóriumok, a konzorciumok (pl.: ClinGen [Clinical Genome Resource, https://clinicalgenome.org]) és a szakértői testületek periodikus frissítéseket hajtanak végre a variánsok újraosztályozására és esetleges átminősítésére, amint további adatok állnak rendelkezésre. Következésképpen az adatbázisok bővülése eredményezheti egy VUS átminősítését patogén vagy valószínűleg patogén variánssá (ezzel a genetikai teszt eredményét pozitívra változtatva), illetve benignus/valószínűleg benignus variánssá történő kategorizálását (53).
Érdemes említést tenni egy esetleges 6. kategóriáról; a másodlagos klinikai relevanciával rendelkező variánsokról. A másodlagos találatok olyan genetikai teszteredmények, amelyek nem kapcsolódnak szorosan, azonban relevánsak a vizsgálatot indokló betegséghez/fenotípushoz, és ismerten betegséget okozó variánsok. E másodlagos találatok jelentésének célja az egészségügyi előnyök biztosítása, egyes betegségek megelőzése vagy előnyösebb kezelése. Az ACMG szerint a teljes exom- és genomszekvenálást végző laboratóriumoknak javasolt bizonyos másodlagos találatokat is riportálni. A 2021-ben frissített ajánlás 73 ilyen gént tartalmaz, amelyek közül 33 kardiovaszkuláris betegséghez kapcsolt gén (54).
A variánsok osztályozása magas szintű szakértelmet igényel és ideális esetben multidiszciplináris megbeszélés keretein belül hajtják végre. A molekuláris genetikai lelet tartalmazza az azonosított kóroki és VUS-génvariánsok listáját, ami magában foglalja a génnevet, az adott variáns azonosításához elengedhetetlen szekvenciaváltozást, annak típusát, az aminosavcserét, a genomi elhelyezkedést, a zigozitást, a variánshoz kapcsolódó betegségeket és a variáns osztályozását klinikai relevancia alapján. Emellett az interpretációban szerepelnek szakirodalmi hivatkozások is, mint például a variánsképződő fehérjére gyakorolt vagy várható hatását vizsgáló in silico, funkcionális és klinikai tanulmányok, amelyek alátámasztják a variánsok klinikai relevanciáját, és egyben osztályozását (55).
Saját tapasztalatok: Komplex kardiogenetikai diagnosztika a Semmelweis Egyetemen
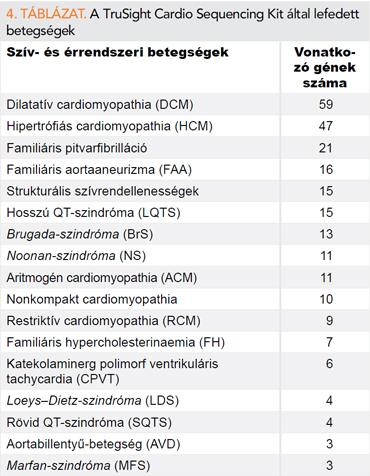
2020-ban került beállításra a Semmelweis Egyetemen egy komplex kardiogenetikai szolgáltatás, amelynek folyamatában az ACMG ajánlása szerint kardiológus, klinikai genetikus, molekuláris biológus, bioinformatikus és patológus is részt vesz (51). Minden vizsgálat előtt klinikai genetikai tanácsadás történik, amelynek során a beteg megismeri a genetikai vizsgálat jelentőségét és lehetséges kimenetelét. A molekuláris analízist NGS segítségével végezzük. Az alkalmazott eljárás (TruSight Cardio Sequencing Kit) 174, korábban örökletes kardiovaszkuláris betegségekkel kapcsolatba hozott gén vizsgálatára alkalmas (4. táblázat). Magába foglalja a kardiológiai betegségek területén részletesen jellemzett, szorosan kapcsolt és ezen betegségekhez asszociált (kimutatták, azonban nem tisztázott a szerepük a szívbetegségek kialakulásában) géneket is (56).
A kapott genetikai eredményt szükséges a klinikai tünetekkel, képalkotó vizsgálatokkal és családi anamnézissel korreláltatni, hogy a molekuláris genetikai lelet a valóban releváns variánsokat tartalmazza. Eddig 102 beteg vizsgálatát végeztük el, amelyek 39%-ában azonosítottunk patogén vagy valószínűleg patogén mutációt. Eredményeink főbb betegcsoportok szerinti bontásban a 2. ábrán láthatóak.

Következtetések
A genetikai tesztelés már elérhető a különböző kardiovaszkuláris megbetegedések vizsgálatára is. Gyakran klinikai és/vagy képalkotó vizsgálatok során megállapított fenotípusos eltérések vagy pozitív családi anamnézis hívja fel a figyelmet genetikai eredetű cardiomyopathiára, aortopathiára vagy aritmiára. A genetikai eredmény és a klinikai diagnózis együttes értékelésével biztosítható a beteg számára a pontos diagnózis, a megfelelő időben alkalmazott optimális kezelés, vagy az ideális életmódbeli és sportolási javaslat.
Támogatás
Innovációs és Technológiai Minisztérium Tématerületi Kiválósági Programja (2020-4.1.1.-TKP2020, TKP2021-NKTA-46, TKP2021-EGA-24, TKP2021-NVA-15), NVKP_16-1-2016-0017 és K20-135076 számú projekt a Nemzeti Kutatási Fejlesztési és Innovációs Alapból biztosított támogatással, az Európai Únió H2020-739593 programja, valamint az Elixir Hungary hálózat.
IRODALOM
1. Venter JC, et al. The sequence of the human genome. Science 2001; 291(5507): 1304–51. https://doi.org/10.1126/science.1058040
2. Mogensen J, et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: a viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur Heart J 2015; 36(22): 1367–70. https://doi.org/10.1093/eurheartj/ehv122
3. Maron BJ, Maron MS, Semsarian. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8): 705–15. https://doi.org/10.1016/j.jacc.2012.02.068
4. Ommen SR, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 2020; 76(25): e159–e240. https://doi.org/10.1016/j.jacc.2020.08.045
5. Csanyi B, et al. Identification of a Novel GLA Gene Mutation, Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype. Int Heart J 2017; 58(3): 454–458. https://doi.org/10.1536/ihj.16-361
6. Jordan E, et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021; 144(1): 7–19.
https://doi.org/10.1161/CIRCULATIONAHA.120.053033
7. Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012; 366(7): 619–28.
https://doi.org/10.1056/NEJMoa1110186
8. Franaszczyk M, et al. Titin Truncating Variants in Dilated Cardiomyopathy – Prevalence and Genotype-Phenotype Correlations. PLoS One 2017; 12(1): e0169007.
https://doi.org/10.1371/journal.pone.0169007
9. Chen SN, et al. Lamin A/C Cardiomyopathy: Implications for Treatment. Curr Cardiol Rep 2019; 21(12): 160.
https://doi.org/10.1007/s11886-019-1224-7
10. Al-Khatib SM, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018; 138(13): e210–e271.
https://doi.org/10.1161/CIR.0000000000000548
11. Pelliccia A, et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J 2021; 42(1): 17–96. https://doi.org/10.1093/eurheartj/ehaa605
12. Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med 2008; 5(5): 258–67. https://doi.org/10.1038/ncpcardio1182
13. Elliott P, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2010; 3(4): 314–22. https://doi.org/10.1161/CIRCGENETICS.110.937805
14. Gallego-Delgado M, et al. Idiopathic Restrictive Cardiomyopathy Is Primarily a Genetic Disease. J Am Coll Cardiol 2016; 67(25): 3021–3. https://doi.org/10.1016/j.jacc.2016.04.024
15. Wechalekar AD, Gillmore JD, HawkinsPN. Systemic amyloidosis. Lancet 2016; 387(10038): 2641–2654.
https://doi.org/10.1016/S0140-6736(15)01274-X
16. Hollerer IA, Bachmann Muckenthaler MU. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica 2017; 102(5): 809–817.
https://doi.org/10.3324/haematol.2016.160432
17. Di Toro A, et al. Myths to debunk: the non-compacted myocardium. Eur Heart J Suppl 2020; 22(Suppl L): L6–L10.
https://doi.org/10.1093/eurheartj/suaa124
18. Arbustini E, et al. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J Am Coll Cardiol 2016; 68(9): 949–66. https://doi.org/10.1016/j.jacc.2016.05.096
19. Arbustini E, et al. Cardiac Phenotypes in Hereditary Muscle Disorders: JACC State–of-the-Art Review. J Am Coll Cardiol 2018; 72(20): 2485–2506. https://doi.org/10.1016/j.jacc.2018.08.2182
20. Loeys BL, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355(8): 788–98.
https://doi.org/10.1056/NEJMoa055695
21. Byers PH, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet 2017; 175(1): 40–47. https://doi.org/10.1002/ajmg.c.31553
22. Frank M, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet 2015;23(12): 1657–64.
https://doi.org/10.1038/ejhg.2015.32
23. Faivre L, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007; 81(3): 454–66. https://doi.org/10.1086/520125
24. Renard M, et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol 2018; 72(6): 605–615. https://doi.org/10.1016/j.jacc.2018.04.089
25. Ackerman MJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011; 8(8): 1308–39.
https://doi.org/10.1016/j.hrthm.2011.05.020
26. Al-Khatib SM, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018; 138(13): e272–e391. https://doi.org/10.1161/CIR.0000000000000549
27. Schwartz PJ, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001; 103(1): 89–95. https://doi.org/10.1161/01.cir.103.1.89
28. Hosseini SM, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence–Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 2018; 138(12): 1195–1205.
https://doi.org/10.1161/CIRCULATIONAHA.118.035070
29. Milman A, et al. Gender differences in patients with Brugada syndrome and arrhythmic events: Data from a survey on arrhythmic events in 678 patients. Heart Rhythm 2018; 15(10): 1457–1465. https://doi.org/10.1016/j.hrthm.2018.06.019
30. Chung FP, et al. A novel method to enhance phenotype, epicardial functional substrates, and ventricular tachyarrhythmia in Brugada syndrome. Heart Rhythm 2017; 14(4): 58–517.
https://doi.org/10.1016/j.hrthm.2017.01.006
31. Sieira JG. Dendramis, and P. Brugada, Pathogenesis and management of Brugada syndrome. Nat Rev Cardiol 2016; 13(12): 744–756. https://doi.org/10.1038/nrcardio.2016.143
32. Al-Khatib SM, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2018; 72(14): e91–e220. https://doi.org/10.1016/j.jacc.2017.10.054
33. Priori SG, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015; 36(41): 2793–2867. https://doi.org/10.1093/eurheartj/ehv316
34. Priori SG, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013; 10(12): 1932–63.
https://doi.org/10.1016/j.hrthm.2013.05.014
35. Priori SG, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlies catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103(2): 196–200.
https://doi.org/10.1161/01.cir.103.2.196
36. Lahat H, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 2001; 69(6): 1378–84.
https://doi.org/10.1086/324565
37. Gyorke I, et al. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 2004; 86(4): 2121–8.
https://doi.org/10.1016/S0006-3495(04)74271-X
38. Priori SG, et al. Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia: JACC Focus Seminar 5/5. J Am Coll Cardiol 2021; 77(20): 2592–2612.
https://doi.org/10.1016/j.jacc.2020.12.073
39. Smith ED, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020; 141(23): 1872–1884.
https://doi.org/10.1161/CIRCULATIONAHA.119.044934
40. Begay RL, et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin Electrophysiol 2018; 4(4): 504–514. https://doi.org/10.1016/j.jacep.2017.12.003
41. Ingles J, et al. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012; 98(8): 625–30. https://doi.org/10.1136/heartjnl-2011-300368
42. Schmidtke JK, Wittkowski, Glaubitz R. NGS-Based genetic testing for heritable cardiovascular diseases. Specific requirements for obtaining informed consent. Mol Cell Probes 2019; 45: 70–78.
https://doi.org/10.1016/j.mcp.2019.04.005
43. Kalayinia S, et al. Next generation sequencing applications for cardiovascular disease. Ann Med 2018. 50(2): 91–109.
https://doi.org/10.1080/07853890.2017.1392595
44. Ronaghi M, et al. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996; 242(1): 84–9. Ronaghi M, et al. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996; 242(1): 84–9.
45. Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem 2009; 55(4): 641–58. https://doi.org/10.1373/clinchem.2008.112789
46. Hategan L, Borbás J, Pálinkás E, Takács ED, Nagy V, Sepp R. Genetic diagnosis in hypertrophic cardiomyopathy: two steps forward, one step back. Cardiologia Hungarica 2021; 51: 109–117.
47. Nagy VA, Tringer A, Lidia H, Csányi B, Pálinkás E, Borbás J, Hegedűs Z, Nagy I, Sepp R. Béta-myozin nehézlánc- és myozin kötő C-fehérje gén kettős mutáció azonosítása malignus megjelenésű hypertrophiás cardiomyopathia hátterében. Cardiologia Hungarica 2019; 49: 431–6.
48. Csányi B, Rudas L, Babik B, Nagy V, Tringer A, Hategan L, Borbás J, Hegedűs Z, Nagy I, Sepp R. Kettős titin és desmoplakin génmutáció igazolása peripartum cardiomyopathiában: a szívtranszplantáción Szegeden átesett beteg genetikai analízise. Cardiologia Hungarica 2020; 50: 132–136.
49. Sepp R, Napolitano C, Pálinkás A, Anastasakis A, Csanádi Z, Priori SG, Schwartz PJ, Forster T. Az első KCNQ1-génmutáció azonosítása hosszú QT-szindrómás magyar betegben. Cardiologia Hungarica 2006; 36: 11–16
50. Sepp R, Nagy V, Sághy L, Napolitano C, Józan-Jilling M, Priori S, Csanády M, Forster T. Az első KCNE1-génmutáció azonosítása magyar hosszú QT-szindrómás betegben. Cardiologia Hungarica 2010; 40: 197–202
51. Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5): 405–24.
52. Richards CS, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008; 10(4): 294–300.
https://doi.org/10.1097/GIM.0b013e31816b5cae
53. Semsarian C, et al. Precision Medicine in Cardiovascular Disease: Genetics and Impact on Phenotypes: JACC Focus Seminar 1/5. J Am Coll Cardiol 2021; 77(20): 2517–2530.
https://doi.org/10.1016/j.jacc.2020.12.071
54. Miller DT, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021; 23(8): 1381–1390.
https://doi.org/10.1038/s41436-021-01172-3
55. Otto CM, Savla JJ, Hisama FM. Cardiogenetics: a primer for the clinical cardiologist. Heart 2020; 106(12): 938–947.
https://doi.org/10.1136/heartjnl-2019-316241
56. Pua CJ, et al. Development of a Comprehensive Sequencing Assay for Inherited Cardiac Condition Genes. J Cardiovasc Transl Res 2016; 9(1): 3–11. https://doi.org/10.1007/s12265-016-9673-5