Late diagnosis of transthyretin amyloidosis causing severe heart failure
█ Case report
DOI: 10.26430/CHUNGARICA.2021.51.2.131
Authors:
Márk László1, Reiber István2, Bajnok László3, Paragh György4
1Békés Megyei Központi Kórház Pándy Kálmán Tagkórház, Kardiológia Osztály, Gyula
2Fejér Megyei Szt. György Egyetemi Oktató Kórház, Székesfehérvár
3Pécsi Tudományegyetem, Klinikai Központ, I. sz. Belgyógyászati Klinika, Endokrinológiai és Anyagcsere Tanszék, Pécs
4Debreceni Egyetem, Belgyógyászati Intézet, Anyagcsere Betegségek Tanszék, Debrecen
Levelezési cím:
Dr. Márk László, Békés Megyei Központi Kórház Pándy Kálmán Tagkórház, Kardiológia Osztály
5700 Gyula, Semmelweis u. 1. E-mail: dr.mark.laszlo@gmail.com
Summary
Late diagnosis of transthyretin amyloidosis causing severe heart failure
Several heart muscle diseases can cause heart failure, the etiological diagnosis of which may serve the basis of targeted, tailored, effective personalized therapy. Such a heart muscle disease is transthyretin amyloidosis (ATTR) which is an infiltrative disorder affecting most frequently the heart and peripheral nerves, due to the accumulation of transthyretin protein. Deposition of mutant transthyretin, due to mutations in the TTR gene encoding for transthyretin, occurs in the familial form of the disease (hATTR), while in senile amyloidosis, wild-type transthyretin accumulates (wtATTR).
We report on a 79-years-old male patient who presented 9 years before because of novel onset atrial fibrillation. Echocardiography at that time showed severe concentric left ventricular hypertrophy which progressed through the years. Laboratory values displayed increased liver enzymes and troponin T levels. He was hospitalized several times because of left- and later right-sided heart failure, with increasing tendency towards hypotension. At last admission he was admitted to us after suffering syncope at home necessitating cardiopulmonary resuscitation.
Transthoracic echocardiography showed severe, concentric biventricular hypertrophy (LV wall thickness 22 mm, RV wall thickness 12 mm), with depressed LV and RV function, apical sparing, and low cardiac output. Taking low voltage, seen on ECG, and other anamnestic data into consideration, the suspicion of ATTR amyloidosis was raised. The patient died after a short observational period due to intractable heart failure. Autopsy and histology revealed systemic amyloidosis, affecting predominantly the heart which was interpreted as wtATTR according to immunohistochemistry, negative TTR gene sequencing and the lack of monoclonal gammopathy.
Our case illustrates the importance of the diagnostic delay which is very frequently encountered in TTR amyloidosis. Features of ATTR which includes the multidisciplinary nature of assessment, specialized diagnostic modalities and unique therapy call for the need of specialized diagnostic centers.
ISSUE: CARDIOLOGIA HUNGARICA | 2021 | VOLUME 51, ISSUE 2
Összefoglaló
Szívelégtelenség hátterében számos szívizombetegség állhat, amelyek etiológiai diagnózisa alapvető feltétele a célzott, személyre szabott, effektív terápiának. Ilyen szívizombetegség a transthyretin-amyloidosis (ATTR), amely a transthyretin-fehérje lerakódása következtében létrejövő szisztémás infiltratív betegség és leggyakrabban a szívet és a perifériás idegrendszert érinti. Örökletes formáját, amelyben mutáns transthyretin rakódik le (hATTR), a transthyretint kódoló TTR-gén mutációi okozzák, míg vad típusú transthyretin akkumulációja esetén szenilis amyloidosisról (wtATTR) beszélünk.
Munkánkban egy 79 éves férfi beteg esetét ismertetjük, akinek első kardiológiai észlelése 9 évvel korábban történt novum pitvarfibrilláció miatt. Echokardiográfiával súlyos koncentrikus balkamra-hipertrófia került leírásra, amely fokozatos progressziót mutatott. Laborjában tartósan emelkedett májenzimértékek és Troponin-T-szint volt látható. Többször került hospitalizációra bal-, majd később már jobbszívfél-elégtelenség tüneteivel, az egyre inkább kifejezett hipotóniahajlam mellett. Legutolsó felvétele otthonában eszméletvesztést követően, reanimáció után történt. Az általunk végzett transthoracalis echokardiográfiás vizsgálat súlyos koncentrikus, mindkét kamrát érintő hipertrófiát (balkamrafal-vastagság 22 mm, jobbkamrafal-vastagság 12 mm) mutatott, csökkent szisztolés bal- és jobbkamra-funkció, emelkedett töltőnyomások és „apical sparing” mellett, alacsony perctérfogat-szindrómával.
Az EKG-n látott low voltage és a részletes anamnesztikus adatok ismeretében a legvalószínűbben ATTR-amyloidosis lehetősége merült fel. Ezt követően a beteg rövid észlelési idő után intraktábilis szívelégtelenség következtében exitált. Kórboncolás során szisztémás, legsúlyosabb mértékben a szívben manifesztálódott amyloidosist véleményeztünk, amely az immunhisztokémiai vizsgálat, a negatív TTR-génszekvenálás eredménye ismeretében és a monoklonális gammopathia hiányában vadtípusú, wtATTR-amyloidosisnak felelt meg.
Esetünk jól illusztrálja az ATTR-amyloidosisban gyakran tapasztalható diagnosztikus késlekedés jelentőségét. Utóbbi a multidiszciplináris szemlélet, a specializált diagnosztika és egyedi terápia okán specializált diagnosztikus központok szükségességére hívja fel a figyelmet.
Bevezetés
Szívelégtelenség hátterében számos szívizombetegség állhat, amelyek etiológiai diagnózisa alapvető feltétele a célzott, személyre szabott, effektív terápiának (1, 2). Az ilyen betegségek többsége balkamra-hipertrófia vagy hipertrófiás cardiomyopathia (HCM) fenokópia formájában jelenik meg, amelyek morfológiailag HCM képét utánozzák, de a létrejövő balkamra-hipertrófia etiológiai és patofiziológiai tényezői alapvetően különböznek a „sarcomer-HCM”-étől. A HCM-fenokópiák öröklődésének módja, a betegség természetes lefolyása és kezelése alapvetően eltér a sarcomermutációk által okozott HCM-es betegekétől, ezért ezekben az esetekben az etiológiai diagnózis rendkívüli nagy jelentőségű. Utóbbi nemcsak a pontos diagnózis felállításához nélkülözhetetlen, de a genetikai tanácsadás, prognosztikus megítélés és a megfelelő klinikai kezelés szempontjából is alapvető fontosságú.
A HCM-fenokópiák egyik leggyakoribb megjelenési formája, a kardiális amyloidosis vagy amyloid CMP annak ellenére egy aluldiagnosztizált betegség, hogy nem ritkán áll a szívelégtelenség hátterében (3, 4). A kardiális amyloidosis két legelterjedtebb formája az immunglobulin könnyűlánc (amyloid light-chain, AL) amyloidosis és a transthyretin amyloidosis (ATTR). Az ATTR-CMP magában foglalja a vad típusú TTR (wild-type, wtATTR-CMP, >az esetek 90%-a), valamint az öröklődő, mutáns típusú TTR- (herediter, hATTR-CMP, az esetek <10%) amyloidosist. A betegség a magyarországi szakirodalomban is részletesen diszkutált (5–7). Jelentőségüket két dolog adja:
- prevalenciájuk jóval magasabb lehet, mint azt gondoljuk, egyes közlemények szerint az ismeretlen eredetű balkamra-hipertrófiával vagy megtartott ejekciós frakcióval járó szívelégtelenséggel (HFpEF), illetve súlyos aortastenosisban szenvedő, aortabillentyű-cserén átesett 65 évnél idősebb betegek 6-16%-ának lehet wtATTR-CMP-je (8–10);
- kezelésükben speciális, etiológiai alapú, randomizált klinikai vizsgálat alapján mortalitás- és morbiditáscsökkentő szer áll rendelkezésünkre (11).
Esetismertetésünkben egy transthyretin-amyloidosis esetét mutatjuk be, amely jól illusztrálja a betegségben gyakran tapasztalható diagnosztikus késlekedés jelentőségét. Utóbbi a multidiszciplináris szemlélet, a specializált diagnosztika és egyedi terápia okán specializált diagnosztikai központok szükségességére hívja fel a figyelmet.
Esetismertetés
Klinikai kórlefolyás
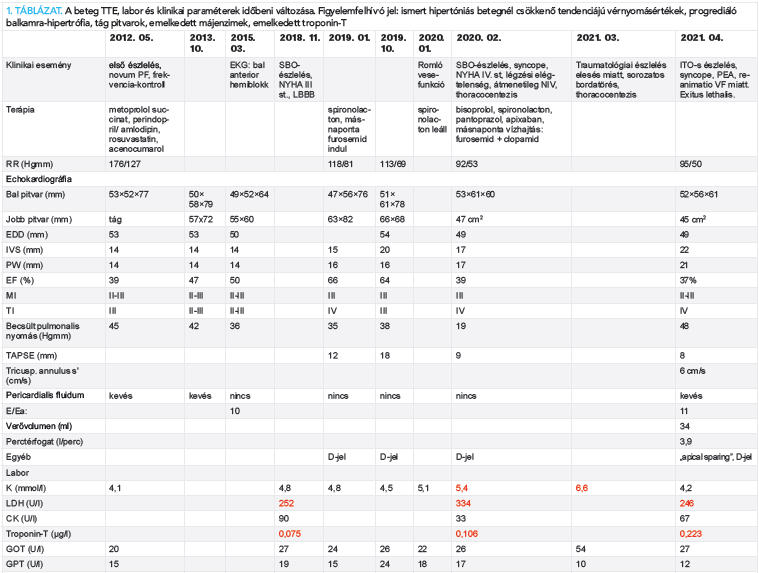
A Szegedi Tudományegyetem AITI Intenzív Osztályán egy 79 éves reanimált, intubált, lélegeztetett, szívelégtelen férfi beteghez kértek szívultrahang-vizsgálatot. A beteg, akinek kronologikus, részletes kórlefolyási adatait, vizsgálati eredményeit, laboreredményeit, echokardiográfiás paramétereit az 1. táblázat tartalmazza, első belgyógyászati észlelése 70 éves korában, 2012-ben történt 1 hete tartó fulladás miatt. Elektrokardiogramon (EKG) 150/min kamrai frekvenciájú, keskeny QRS-ű, novum pitvarfibrilláció, szabályos repolarizáció volt látható. Transthoracalis echokardiográfiával (TTE) tág pitvarok, mérsékelten csökkent balkamra-funkció, közepes fokú mitralis és tricuspidalis regurgitáció került leírásra. Frekvenciakontrollra törekedtek.
Laborjában, normál tartományban lévő paraméterek, euthyreosis volt észlelhető. Terápiaként metoprolol-succinat, perindopril/amlodipin, rosuvastatin, acenocumarol indult. Ezt követően évekig gondozták, csökkent terhelhetőség, vitiumok miatt. Észlelése alatt több alkalommal került sor sürgősségi, kardiológiai osztályos felvételre NYHA III-IV. stádiumú, mindkét szívfél dekompenzációjával járó, hipotenzív akut szívelégtelenség miatt, amely átmeneti noninvazív lélegeztetést, illetve toracocentézist is igényelt. EKG-n bal anterior hemiblokk, majd LBBB jelent meg. TTE-n a balkamra-hipertrófia progressziója volt észlelhető, kifejezett jobbszívfél-terhelés, diasztolés D-jel kialakulásával, majd a balkamra-funkció csökkenésével, tág pitvarokkal, súlyos mitralis és tricuspidalis regurgitációval. Jobbszívfél-elégtelenség hátterében a pulmonológiai okot kizárták, neurológiai szakvizsgálat gócjelet, izomgyengeséget nem talált.
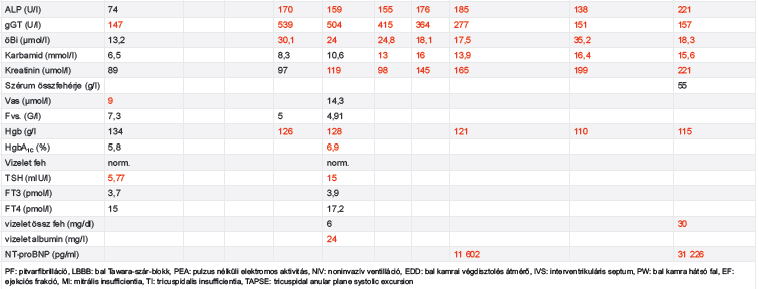
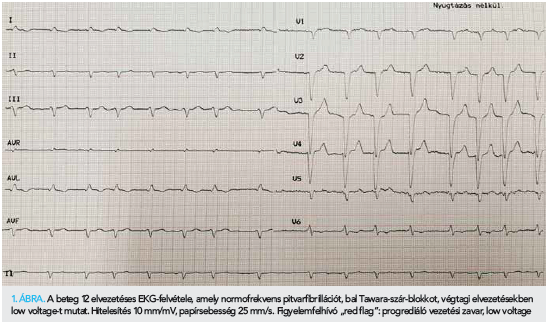
Laborjaiban fokozatosan kialakuló vese- és májfunkció-romlás, anémia volt megfigyelhető. 79 éves korában, 2021 áprilisában, otthonában eszméletét vesztette. Kiérkező mentőszolgálat iniciális ritmusként pulzus nélküli elektromos aktivitást észlelt. Reanimációt kezdtek, több alkalommal kamrafibrilláció miatt sokk leadására is szükség volt. Urgens koronarográfia az epikardiális ereken szűkületet nem igazolt. Ezt követően intubálva és lélegeztetve Intenzív osztályra került. A keringésleállás okának tisztázása érdekében koponya, nyak és mellkas CT-angio vizsgálat történt, amely jelentős fokú jobb oldali hydrothraxot és következményes teljes atelectasiát igazolt. Ismételt mellkascsövezés során 3000 ml serosus pleurális folyadékot bocsátottak le. Az EKG-n low voltage mutatkozott (1. ábra). A kritikus állapotban lévő beteg kiterjesztett TTE-vizsgálata során alacsony perctérfogat-szindróma, súlyos koncentrikus, mindkét kamrát érintő hipertrófia (balkamrafal-vastagság 22 mm, jobb kamra szabad falának vastagsága 12 mm), csökkent szisztolés bal- és jobbkamra-funkció, emelkedett töltőnyomások és „apical sparing” igazolódott (2. ábra, A–D panelek).
Fentiek alapján kardiális amyloidosist véleményeztünk, tekintettel a hosszú évek óta fokozatosan progrediáló SZE-re, senilis amyloidosist tartottuk a legvalószínűbbnek. Laborjában hypercalcaemia nem igazolódott, szérum összfehérje: 51 µmol/l, albumin: 30 µmol/l volt. 24 órás vizeletösszfehérje-ürítés nem volt lényegesen emelkedett. Szérum elektroforézis monoklonális gammopathiát nem igazolt. A beteg az intenzív osztályos kezelés 3. napján intraktábilis szívelégtelenség tünetei között exitált.
Kórbonctani, kórszövettani és genetikai vizsgálat
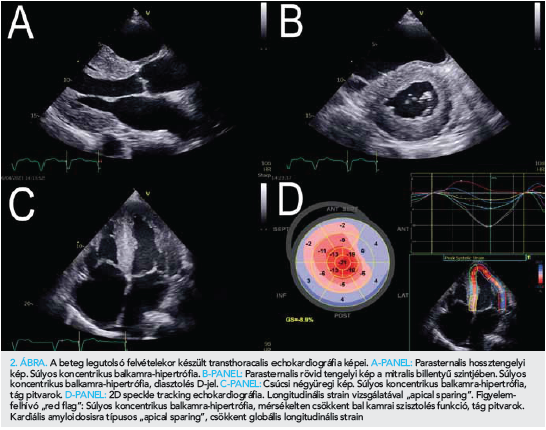
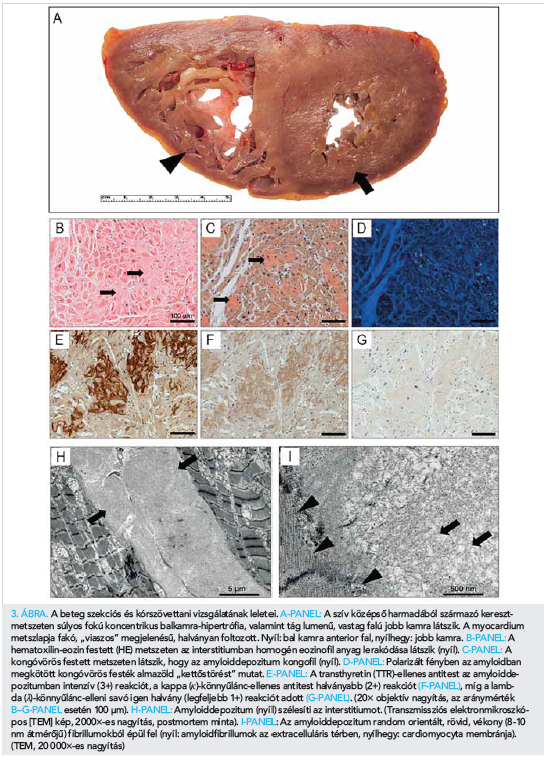
A boncolás során 920 g tömegű, extrém fokban megnagyobbodott szívet észleltünk. A szívüregek vizsgálata súlyos koncentrikus balkamra (BK)-hipertrófiát (maximális BK-falvastagság 24 mm), súlyos dilatatív jobbkamra (JK)-hipertrófiát (maximális JK-falvastagság 12 mm), tág bal- és jobb pitvart igazolt.
A myocardium gumiszerűen tömött, metszlapja fakó, „viaszos” megjelenésű, halványan foltozott volt (3. ábra, A-panel). A szövettani vizsgálat során a valamennyi jobb- és bal kamrai kimetszésben a myocardium súlyos, diffúz interstitialis kiszélesedését észleltük (3. ábra, B-panel). Az interstitiumban lerakódott anyag amyloiddepozícióra utalóan kongóvörös festéssel intenzív vörös színű, polarizált fényben almazöld „kettőstörést” mutatott (3. ábra, C- és D-panel).
A formalin-fixált, paraffinba ágyazott mintákon immunhisztokémiai vizsgálattal a transthyretin (TTR)-ellenes savó (DAKO, poliklonális nyúlantitest 1:1600) az amyloiddepozitumban intenzív (3+) reakciót (3. ábra, E-panel), a kappa (k)-könnyűlánc-ellenes savó (Celmarque, L1C1 monoklonális egérantitest) 2+ (3. ábra, F-panel), a lambda (λ)-könnyűlánc-ellenes antitest (Labvision, HP6054 monoclonalis egérantitest) legfeljebb 1+ reakciót adott (3. ábra, G-panel). A myocardiumon kívül amyloidot találtunk a szubepikardiális zsírszövetben és a koszorúerek tunica mediájában.
Extrakardiálisan az amyloid középnagy- és kisartériák falában és kapillárisok környezetében volt jellemző. A glomerulusokban, a lépben és a májban nem észleltünk amyloidlerakódást. A csontokon lítikus léziót nem észleltünk. A thoracalis csigolyából származó csontvelőben szembetűnő plazmasejt-szaporulat nem állt fenn. A jobb kamra kifolyó traktusának septalis felszínéből kimetszett parányi szubendokardiális mintát glutáraldehidben fixáltuk, amelyből műgyantás beágyazást és osmiumos fixálást követően transzmissziós elektronmikroszkópos vizsgálatot végeztünk (JEOL-1400 TEM). Az interstitiumban véletlenszerűen rendeződött, periodicitás-mentes fibrillumokból felépülő anyag lerakódását észleltük (3. ábra, H és I panel). A fibrillumok átmérőjét 9,9±1,7 nm-nek (átlag ±SD) mértük (30 fibrillumot mértünk, keresztmetszeti síkban, 40 000×-es nagyításon).
A beteg hozzátartozójának tájékoztatott beleegyezése után a beteg perifériás vérmintájából genetikai vizsgálatot végeztünk a transthyretin (TTR)-gén teljes kódoló szakaszának direkt szekvenálásával. A szekvenciaanalízis TTR-génmutációt nem igazolt.
Megbeszélés
Esetünkben a súlyos, végső soron halálhoz vezető szívelégtelenség hátterében szisztémás, legsúlyosabb mértékben a szívben manifesztálódott amyloidosis állt. Az összkép leginkább vadtípusú, wtATTR-amyloidosisnak felel meg; tekintettel arra, hogy a szérum elektroforézis AL-amyloidosishoz társuló monoklonális gammopathiat nem igazolt, a kórszövettani vizsgálatkor a szívben leírt kappa és lambda könnyűlánc-festődés aspecifikus festődésként értékelhető. A tisztán ATTR-amyloidosis, vagy egyéb kevert amyloidosis 100%-os bizonyossággal való elkülönítése lézer-mikrodisszekciós tömegspektrometria-alapú (LDMS) amyloidtipizálás alapján lett volna lehetséges, azonban ez nem állt rendelkezésünkre. Az immunhisztokémiával, illetve az LDMS-sel meghatározott amyloidtípus között az esetek 9%-ában találtak eltérést Rezk és munkatársai (12). A fenti esetben specifikus etiológia klinikai gyanúja csak végstádiumú állapotban, az intenzív osztályon merült fel, definitív diagnózis pedig csak postmortem született.
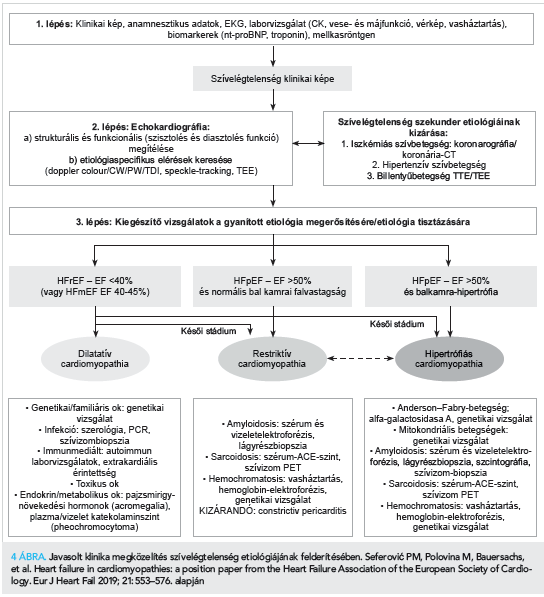
Az eset jól illusztrálja az etiológiai diagnózis felállításának fontosságát olyan kórképekben, ahol a szívelégtelenség általános, tünetorientált terápiája nem elégséges, és specifikus, új terápiás lehetőségek állnak rendelkezésünkre. Ezekben az esetekben mindenféleképpen olyan megközelítés szükséges, amely multimodális képalkotó és egyéb diagnosztikai módszereket kihasználva a pontos etiológia tisztázását teszi lehetővé. Ez érvényes mind az infiltratív kórképekben (mint a kardiális amyloidosis vagy sarcoidosis), tárolási betegségekben, öröklődő szívizombetegségekben, vagy a csökkent ejekciós frakcióval járó szívelégtelenség bizonyos eseteiben (4. ábra).
Ezekre a betegségekre érdemes gyakrabban gondolni, a figyelemfelhívó jeleket „red flag”-et észrevenni, a beteget pedig ezirányú tapasztalattal rendelkező centrumba irányítani (2). Az ATTR-CMP gyanúját a jellemző kardiális eltérések mellett meglévő nem specifikus jelek kelthetik fel. Ilyenek közé tartozhat az idősebb kor (>65 év), a férfinem, a gerincszűkület jelenléte, a carpalis alagútszindróma és a bicepszínszakadás. Kardiális eltérések közé a HFpEF jelenléte egyidejű jobbszívfél-elégtelenséggel, a falvastagság és az EKG-feszültségek közötti diszkonkordancia, a „low-flow, low-gradient” aortastenosis jelenléte, a diffúz késői kontraszthalmozás jelenléte szív-MRI-vizsgálat során, aránytalanul magas natriuretikus peptidértékek és tartósan emelkedett troponinszintek ACS hiányában (4, 13, 14).
Az amyloidosis, és köztük a ATTR-amyloidosis képalkotó diagnosztikáját az echokardiográfia, szív-MR és radionukleotid szcintigráfia vonatkozásában egy nem régi kiváló közlemény foglalta össze a Cardiologia Hungarica hasábjain (7). Monoklonális (AL) fehérje hiányában a technéciummal jelölt pirofoszfát (99mTc-PYP), a 3,3-difoszfono-1, a 2 propanodikarboxilsav (DPD) vagy a hidroxil-metilén-difoszfonát (HMDP) radionuklid szcintigráfia specifikussága és pozitív prediktív értéke az ATTR-CMP-re 100%, ezzel szemben a szív-MRI pozitív prediktív értéke csak 80%. Az ATTR-CMP végzett szívizom-biopszia szenzitivitása és specificitása 100%. Fentieknek megfelelően az ATTR-CMP igazolására a technéciummal jelölt szív radionuklid szcintigráfia minden olyan betegnél ajánlott, akinél nagy a gyanú az ATTR-CMP-t illetően, a monoklonális (AL) fehérje kizárása után. Genetikai vizsgálatot kell végezni az ATTR-CMP örökletes formájának diagnosztizálására, ha a csontszcintigráfia pozitív, és AL-fehérje hiányzik. Szív- vagy extrakardiális biopsziák ajánlottak, ha a noninvazív diagnosztika nem teszi lehetővé a szívamiloidosis (típusának) határozott diagnózisát. A wtATTR-CMP diagnosztizálása érdekében ajánlott először kizárni az örökletes formát genetikai vizsgálattal.
Az amyloidosis, így az ATTR-amyloidosis terápiájában központi szerepet játszik az euvolémia fenntartása, amely a restriktív diasztolés diszfunkció miatt jelentős kihívást jelent. Ha szívelégtelenség-tünetek jelentkeznek, kacsdiuretikumok és aldoszteronantagonisták kombinációja adható, de az ortosztatikus hipotenzió-intoleranciát okozhat. A hipotenzió miatt előfordulhat, hogy a BBs, ACE-Is, ARB- vagy ARNI-kezelés nem tolerálható jól. Kerülni kell a kalciumantagonistákat, mivel súlyos hipotenziót és fáradtságot okozhatnak, vagy amyloidkomplexeket képezhetnek. Amennyiben pitvarfibrilláció jelentkezik, a PF megerősítését követően az amyloid CMP-ben szenvedő betegeknél antikoaguláns terápiát kell kezdeni.
Az AL-CMP kezelésének alapja a háttérben álló hematológiai alapbetegség kezelése kemoterápiával vagy autológ-őssejt-transzplantációval, amelynek sikere határozza meg az AL-CMP prognózisát.
Az ATTR-CMP specifikus kezelésének alapja a transthyretin stabilizációja és termelésének csökkentése. Az ATTR-ACT (Transthyretin Amyloidosis Cardiomyopathy Clinical Trial) vizsgálat eredményei szerint a biopsziával bizonyított, hATTR és wtATTR-CMP-ben a tafamadis az összhalálozás és a CV-hospitalizáció jelentős csökkenését eredményezte, főként azoknál a betegeknél, akik a vizsgálat megkezdésekor NYHA I. és II. funkcionális stádiumban voltak. A funkcionális javulás 6 hónapon belül következett be, míg a mortalitás közel 2 év után kezdett csökkeni (11). További terápiás lehetőség az intravénás patisiran (16), egy kis RNS-interferens molekula, vagy szubkután inotersen, TTR elleni antiszensz oligonukleotid alkalmazása, azokban a betegekben, akikben kombinált hATTR-polyneuropathia és -CMP áll fenn. A diflunisal off-label alkalmazása a wtATTR-CMP-ben protonpumpa-gátlóval kombinálva is felmerülhet. A máj- és/vagy szívtranszplantáció csak a családi ATTR-CMP végstádiumú betegségében merül fel.
Következtetések
Összefoglalva, esetünk jól illusztrálja az ATTR-amyloidosisban gyakran tapasztalható diagnosztikus késlekedés jelentőségét. Utóbbi a multidiszciplináris szemlélet, a specializált diagnosztika és egyedi terápia okán specializált diagnosztikus központok szükségességére hívja fel a figyelmet.
Köszönetnyilvánítás
A munka a „Ritka betegségek patogenezisének kutatása, új diagnosztikai és terápiás eljárásokat megalapozó fejlesztések” (GINOP-2.3.2-15-2016-00039), az „Életet veSzélyezTető Akut megbetegedések súlYossági és hAlálozási mutatóinak jaVítása transzlációs orvostudományi mEgközelítésben – STAY ALIVE” (GINOP-2.3.2-15-2016-00048) és a Szegedi Tudományegyetem ÁOK Kari Kutatási Alap „Hetényi Géza” pályázatának támogatásával készült.
Irodalom
1. Seferović PM, Polovina M, Bauersachs, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2019; 21: 553–576. https://doi.org/10.1002/ejhf.1461
2. Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 1448–1458.
3. Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med (Lond) 2018; 18(Suppl 2): s30–s35.
4. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. Jd Am Coll Cardiol 2019; 73(22): 2872–2891.
5. Hategan L, Csányi B, Nagy V, et al. Transthyretin génmutáció azonosítása hipertrófiás cardiomyopathia képében megjelenő amyloidosisban. Cardiologia Hungarica 2016; 46: 225–230.
6. Pozsonyi Z, Vágó H, Tóth A, et al. Szívamyloidosis diagnosztikája és kezelése: tapasztalataink és irodalmi áttekintés. Cardiologia Hungarica 2016; 46: 292–300.
7. Pozsonyi Z, Peskó G, Czibor S, et al. A képalkotó vizsgálatok szerepe szívamyloidosis kivizsgálásában. Cardiologia Hungarica 2021; 51: 23–32. https://doi.org/10.26430/CHUNGARICA.2021.51.1.23
8. Ternacle J, Krapf L, Mohty D, Magne J, Nguyen A, Galat A, et al. Aortic Stenosis and Cardiac Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol 2019; 74(21): 2638–51.
9. Russo D, Musumeci MB, Volpe M. The neglected issue of cardiac amyloidosis in trials on heart failure with preserved ejection fraction in the elderly. Eur J Heart Fail 2020; 22(9): 1740–1.
10. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36(38): 2585–94.
11. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018; 379: 1007–1016.
https://doi.org/10.1056/NEJMoa1805689
12. Rezk T, Gilbertson JA, Mangione PP, et al. The complementary role of histology and proteomics for diagnosis and typing of systemic amyloidosis. J Pathol Clin Res July 2019; 5: 145–153.
https://doi.org/10.1002/cjp2.126
13. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012; 126: 1286–1300.
14. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013; 8: 31.
15. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016; 133(24): 2404–12.
16. Solomon SD, Adams D, Kristen A, Grogan M, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019; 139(4): 431–443.